
Abdul Sattar Larik
Not Available
Zahoor Ahmed Soomro
Not Available
Asian Journal of Plant Sciences
Year: 2003 | Volume: 2 | Issue: 7 | Page No.: 551-560







ABSTRACT
Significant and far reaching developments are constantly taking place in the new emerging field of genomics, the study of gene structure and function. Part of the need for genomics capabilities involves the rapidly emerging field of bioinformatics, which involves obtaining, storing and analysing information derived from studying biological systems. Crucial to bioinformatic analysis are computer algorithms that can compare newly isolated genes with databases containing genetic sequence of known function. A gene product is the key to the understanding of the intricate biological phenomenon from simple unicellular organisms to the incredibly complex multicellular organisms. This review places in contemporary context the new information on gene structure such as the role of DNA in information storage, coding of genetic information, flow of information from DNA to protein, the satellite DNA’s, information from RNA. Gene function is critically traced through the flow of information from DNA to RNA; the expression of functional products in prokaryotic and eukaryotic genomes, new proteomic approaches to gene expression/function, genetrap database, feed forward loop (FFL) system, extensive information through cDNA libraries. Informational implications of gene function in eukaryotes include: the nuclear DNA reversion, feedback from nucleus to cytoplasm, gene activity in coupling and repulsion phases, branching enzymes and the ectopic expression of gene and aberrant transcripts. In addition to the academic goals of perceiving gene structure and functions, there is great potential for agricultural and medicinal applications of functional data in the perceiving of plant and human diseases for pragmatic remedies.
PDF Abstract XML References Citation
How to cite this article
Abdul Sattar Larik and Zahoor Ahmed Soomro, 2003. Elucidation of Gene Structure and Function of Prokaryotes and Eukaryotes Through DNA Information Technology. Asian Journal of Plant Sciences, 2: 551-560.
DOI: 10.3923/ajps.2003.551.560
URL: https://scialert.net/abstract/?doi=ajps.2003.551.560
DOI: 10.3923/ajps.2003.551.560
URL: https://scialert.net/abstract/?doi=ajps.2003.551.560
Gene concept and definitions: Starting with Gregor Mendel (1866) the concept of gene has undergone continual revision during last 136 years. Classical genetics teaches that genes are units of inheritance localized in chromosomes, that linked genes are recombinable by crossing over, that an individual gene may be capable of being modified in several ways through mutation, and that genes somehow perform specific functions. There is now strong evidence that DNA rather than protein carries primary genetic information. Later, Watson and Crick (1953a, b) regarded the precise sequence of the bases as the code which carried the genetical information. Gamow (1954) produced the idea that the linear sequence of nucleotides in nucleic acid was responsible for determining the linear sequence of amino acids in the polypeptide chain of protein molecules. Benzer made an enormous advance in delineating the physical extent of the gene when he defined a cistron as a unit of genetic function. The gene was thus subdivided into regulatory regions and structural regions. This subdivision was accelerated with the timely discovery of introns and exons. Introns are the shortest DNA segment that are transcribed but later removed from mRNA by molecular excision before translation, whereas the coding DNA outside the intron is composed of exons Watson et al. (1987).
Benzer (1957) defined cistron by a comparison of the phenotype of CIS and TRANS configuration of a pair of mutations. Thus by this definition a cistron would be a segment of hereditary material within which the cis configuration producing normal phenotype and the trans configuration producing a mutant phenotype. This equates the cistron with the gene defined as a unit of function, responsible for specifying a particular peptide chain. Benzer further proposed the term recon for the recombination and muton for unit of mutation. Recon is the smallest unit that is interchangeable by genetic recombination and the muton is the smallest unit when altered, produces the mutant form of an organism. From this definition it is clear that gene is a unit of function, of recombination as well as of mutation. Benzer’s term cistron can be retained as functional unit of hereditary material as pointed by Fincham (1959) provided the definition of a cistron is modified in such a way that the two mutations in the trans configuration in a heterokaryon or diploid will give normal enzymes. However, if they are in different cistron they will produce abnormal enzyme.
Thus, according to the contemporary concept, a cistron is a part of hereditary material that specifies the amino acid sequence in one polypeptide. It can direct synthesis of protein under precise and appropriate regulatory control. Since, gene is a linear structure with numerous sites of mutation within the functional unit, the mutations cause changes in the amino acid sequence in specific polypeptides. Moreover, there is an exact point by point relationship between the position of the mutation in the gene and the position of the amino acid substitution in the polypeptide. Benzer (1962) suggested that a sequence of three consecutive nucleotides was responsible for each amino acid and that the nucleotide sequence was read at single nucleotide interval. In other words, if a segment of a nucleic acid is presented by the letters ABC ACD where A-D specify the four individual nucleotides, the triplet ABC would represent one amino acid, BCA the next, CAC the third and so on.
Gene structure: Gene is the region of DNA that controls discrete hereditary characteristics. On the other hand, DNA is composed of four basic molecules called nucleotides, which are identical, except that each contains a different nitrogen base, which contains phosphate, sugar and one of the four bases. When phosphate is not present, the base and the deoxyribose form a nucleoside rather than a nucleotide. The four bases are adenine, guanine, cytosine, uracil and thymine. The two of the bases, adenine and guanine are similar in structure and are called purines consisting of two rings. The other two bases cytosine and thymine, also are similar and are called pyrimidines consisting of a single ring. The mystery of DNA structure was investigated by many scientists in the early 1950’s but it was eventually solved by Watson and Crick (1953a). The basic building block of DNA was known to be nucleotide consisting of the five-carbon sugar deoxyribose to which one phosphate is esterified at the 5´ position of sugar ring and one nitrogenous base is attached at the 1´ site. There are two types of bases, the smaller pyrimidines and the larger purines. The nucleotides were known to be covalently linked to one another to form a linear polymer or strand, with a backbone compound of alternating sugar and phosphate groups joined by 3´-5´-phosphodiester bond. A nucleotide has a polarized structure: one edge where the phosphate is located is called 5´ end (five prime end) while the other edge is called 3´ end. DNA on the other hand, was thought to be composed of a monotonous repeat of its four nucleotide building blocks (e.g. ATGCATGCATGC--) which was called tetranucleotide theory. If the tetranucleotide theory was correct, each of the four bases should be present in 25 percent of the total number. Chargaff (1950) found that the ratio of the four components bases were quite variable from one type of organism to another, often very different from 1:1:1:1 ratio predicted by the tetranucleotide theory. For example, the A:G ratio of the DNA of a Tubercle bacillus was 0.4, while the A:G ratio of the human DNA was 1.56. It made no difference which tissue was used as the source of DNA, the base composition remained constant for that species. Amid this great variability in the base composition of different DNA’s, an important numerical relationship was discovered. The number of purines always equalled the number of pyrimidines in a given sample of DNA. More specifically, the number of adenines always equalled the number of thymines, and the number of guanines always equalled the number of cytosines. In other words, Chargaff discovered the following rules of DNA base composition:
| [A] = [T] [G] = [C] [A] + [T] ≠ [G] + [C] |
Role of DNA in information storage: The work of Chargaff and his colleagues on the base composition of DNA shattered the notion that DNA was a molecule consisting of a simple repetitive series of nucleotides. This finding awakened researchers to the possibility that DNA might have the properties necessary to fulfil a role in information storage.
DNA is a helical molecule consisting of two chains of nucleotides running in opposite directions with their backbones on the outside and the nitrogenous bases facing inwards. Each helix is a chain of nucleotides held together by phosphodiester bond, in which phosphate group forms a bridge between –OH group on two adjacent sugar residues. The two helices are held together by hydrogen bonds, in which two electro negative atoms “share” a proton, between the bases. Hydrogen bonds occur between hydrogen atoms with a small positive charges and acceptor atoms with a small negative charge (Fig. 1).
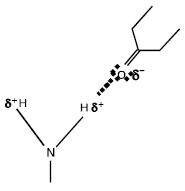 | |
| Fig. 1: | The Hydrogen bonds |
Each hydrogen atom in the NH2 group is slightly positive (δ+) because the nitrogen atom tends to attract the electrons involved in the N-H bond, thereby leaving the hydrogen atom slightly short of electrons. The oxygen atom has six unbounded electrons in its outer shell, making it slightly negative (δ-). A hydrogen bond forms between one H and the O. Hydrogen bonds are quite weak, but this weakness plays an important role in the function of the DNA molecule in heredity. However, the hydrogen bond is much stronger if the participation atoms are “pointing at each other” in the ideal orientations.
Coding of genetic information: Genetic information is encoded in the specific linear sequence of nucleotides that make up the strand. The Watson-Crick model of DNA structure suggested a mechanism of replication that included strand separation and the use of each strand as a template that directed the order of nucleotide assembly during construction of the complementary strand. The model of DNA depicted B-DNA, which is one of several right handed forms. B-DNA contrasts most markedly with Z-DNA, which takes the form of a left handed, helix 18 Å in diameter containing 12 base pairs per turn Rich et al. (1984) in which the backbone assumes a zigzag conformation. The DNA molecule in relaxed state possesses 10 base pairs per turn of the helix. DNA found within a cell tends to be underwound (contains a greater number of base pairs per turn) and is said to be negatively supercoiled, a condition that tends to facilitate the separation of strands that occur during replication and transcription.
The satellite DNA: The supercoiled state of DNA is altered by topoisomerases, enzymes that are able to cut, rearrange and reseal DNA strand Hsieh (1992) when DNA fragments from eukaryotic cells are allowed to reanneal, show three distinct classes of DNA sequences i.e. satellite DNA’s (DNA sequences that are repeated in great number, situated at the centromeres of the chromosomes), minisatellite DNAs and microsatellite DNAs. Each base pair consists of one purine base and pyrimidine base, paired according to the following rule: G pairs with C, and A pairs with T. Such pairing would account for the (A+G) = (T+C). However, G-C pair has three hydrogen bonds, whereas the A-T pair has only two. Therefore, DNA containing many G-C pairs would be more stable than DNA containing many A-T pairs.
Elucidation of the structure of DNA caused a lot of excitement in genetics for two basic reasons. First the structure suggests an obvious way in which the molecule can be duplicated, or replicated, since each base can specify its complementary base by hydrogen bonding. Second, the structure suggests that perhaps the sequence of nucleotide pairs in DNA is dictating the sequence of amino acids in the protein organized by that gene. In other words, some sort of genetic code may write information in DNA as a sequence of nucleotide pairs and then translate it into a different language of amino acid sequence in protein.
Flow of information from DNA to protein: Once the structure of DNA was established, it was evident that the sequence of amino acids in a polypeptide was specified by the nucleotides in the DNA of a gene. It seemed very unlikely that the DNA could serve as direct, physical template for the assembly of a protein. Instead it was widely assumed that the information stored in the nucleotide sequence was present in some type of genetic code. With the discovery of messenger RNA as an intermediate in the flow of information from DNA to protein, attention turned to the manner in which a sequence “written” in a ribonucleotide “alphabet” would be able to code for a sequence “written” in a very different alphabet consisting of amino acids.
Information from RNA: The first clear evidence that RNA has the capacity to carry information about protein structure came from tobacco mosaic virus (TMV). This virus contains no DNA. It is composed of RNA surrounded by a hallow cylinder of protein measuring 300 nm in length and 15 nm in external diameter. Gierer and Schramm (1956) separated the RNA and protein and showed that the RNA by itself is ineffective, although to much lesser degree (0.1%) compared with the intact virus. Fraenckle-Conrat (1956) mixed the RNA of one strains and the proteins of another and produced several very active chimeras. The serological characters of these mixed virus preparations were those of the virus supplying the protein, but all the characters of the progeny were those of the virus supplying the RNA, suggesting that ribose nucleic acid (RNA) is the sole bridge between generations. Thus RNA, too, must be capable of carrying genetic information.
Fraenkel-Conrat and Singer (1957) investigated this remarkable discovery in more detail in Nicotiana tabacum. In view of clear evidence that RNA can carry genetic information about protein structure it is not surprising that the structure of RNA appears to be quite similar to that of DNA. The essential differences are that the sugar is D-ribose instead of 2-deoxy-D-ribose and one of the pyrimidines is Uracil in place of Thimine of DNA. In other three bases (adenine, guanine and cytosine) are the same. Nucleotide sequence in DNA is first described into messenger RNA and then translated into amino-acid.
Gene function: Inside the eukaryotic nucleus some genes are active more or less constantly, but other have to be turned on and off to suit the needs of the cell or the organism. The signal to activate a gene may come from outside the cell, for example a substance such as steroid hormone. Alternatively, the signal may come from within the cell, for example from a special regulatory genes whose job is to turn other genes on and off. The regulatory substances bind to a special region of the gene and initiate the synthesis of a copies of the genes DNA. This DNA is transcriped into an messenger RNA (mRNA) molecule, which is then translated during protein synthesis. Translation requires transfer RNA (tRNA) and ribosomes. Eukaryotic genes are activated by two Cis-acting controlling sequences –promoters and enhancers- which are recognised by trans- acting proteins. The trans-acting proteins allow the RNA polymerase to initiate transcription and to achieve maximal rates of transcription. In eukaryotes, the initial RNA transcript is processed in several ways to generate the final mRNA. Many eukaryotic genes contains non-coding regions called introns, that interrupt the normal genes coding sequence.
Flow of information from DNA to RNA: The mRNA molecules pass out through the nuclear pores into the cytoplasm and here the information in the sequence of mRNA is translated into an amino acid sequence in protein by a complex translational appartus (Fig. 2).
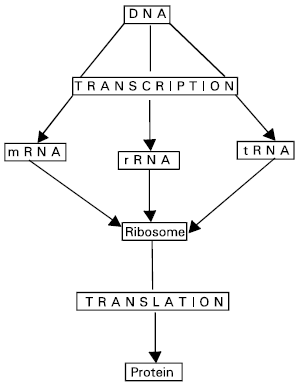 | |
| Fig. 2: | A simplified view of information flow involving DNA, RNA and protein within cell |
Each gene code for a separate protein, each with specific functions either within the cell or for export to other parts of the organism. Proteins are the most important manifestations of forms in living organism either as a structural component or as a regulator of the body chemistry. The flow of information from DNA to RNA to protein is the central dogma of molecular biology.
Expression of functional products in prokaryotic and eukaryotic genomes: Gene is the region of DNA that controls a discrete hereditary characteristics, usually corresponding to a single protein or RNA. This definition includes the entire functional unit, encompassing coding DNA sequences, noncoding regulatory DNA sequences and introns. In molecular terms a gene can be defined as segment of DNA that is expressed to yield a functional product, which may be either an RNA (e.g. rRNA and tRNA) or a polypeptide. Gene vary greatly in size from less than 100 base pairs to several thousand base pairs. Similarly, great variation is also found in the number of genes from one organism to the other. In bacterial genome, most of the DNA encodes protein. For example, the genome of E. coli is approximately 4.6 x 106 base pairs long and contains 4288 genes, with nearly 90% of the DNA used as protein–coding sequences. In Saccharomyces cerevisiae the average gene spans about 2000 base pairs, and approximately 70% of the yeast genome is used as protein- coding sequence, specifying a total of about 6000 proteins. Prokaryotic genome contains a single chromosome, with circular DNA molecule, whereas eukaryotic genomes are composed of multiple chromosomes, each containing a linear molecule of DNA. Although the numbers and sizes of chromosomes vary considerably between different species their basic structure is the same in all eukaryotes. The DNA of eukaryotic cells is tightly bound to small basic protein (histones) that package the DNA in an orderly way in the cell nucleus. The complexes between eukaryotic DNA and protein are called chromatin. The basic structural unit of chromatin is called nucleosome. Chromatin may be functional or non-functional. Heterochromatin is inactive chromatin, whereas euchromatin is actively involved in RNA transcription.
Changing interpretations of gene function: The first meaningful insight into gene function was made by Garrod (1902) who concluded that persons suffering from inherited metabolic disease were missing specific enzymes. Later, Beadle and Tatum (1941) were able to produce mutations in the genes of Neurospora and identifying the specific metabolic reactions that were affected. These studied led to the concept of “one-gene-one-enzyme” and subsequently to more refined version of one-cistron-one-polypeptide hypothesis. According to Beadle and Tatum a biosynthetic pathway might have four steps, where 1 is the starting material and 5 is the final product.
Each step is catalyzed by an enzyme: A, B, C or D. In turn, each enzyme is specified by a particular gene. This would imply that gene a specifies enzyme A, gene b specifies enzyme B, and so on. Therefore, if we inactivate the gene responsible for an enzyme, we initiate one required step and the pathway is interrupted. Following diagram shows that enzyme B is eliminated due to mutation in gene b.
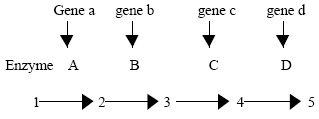 |
Since, the cell cannot carry out the reaction that converts compound 2 to compound 3, it is blocked at compound 2, and can not go further. The model one-gene-one enzyme hypothesis provided the first exciting insight into the function of genes. Genes some how were responsible for the function of enzymes and each gene apparently controlled one specific enzyme. The hypothesis can be summarized as under: (I) biochemical reactions in vivo occur as a series of discrete stepwise reactions (ii) each reaction is specifically catalyzed by a single enzyme (iii) each enzyme is specified by a single gene. The gene is a unit of function which is equivalent to the cistron and that can be defined experimentally by a cis-trans complementation test. The cistron is a region of the genetic material that codes for one polypeptide chain. Therefore, one-gene-one-enzyme hypothesis be referred more precisely as the one-cistron-one-polypeptide hypothesis, thereby emphasizing that cistron can code for proteins other than enzyme.
By 1964, it become clear that the DNA associated with the nucleus contains genes which direct the synthesis of ribosomal RNA Perry (1964). This was conclusively proved in case of mammalian cells by McConkey and Hopkins (1964). The one-cistron-one-polypeptide hypothesis is strongly supported by the studies of Schwartz (1965) dealing with esterase mutations in maize. His evidence that isozymes characteristic of an allelic series arise as a result of charge differences serves to clarify this and similar situations which initially appeared to be in potential conflict with this hypothesis in its simplest form.
New proteomic approaches to gene expression/function: Full-length cDNA sequencing allows unique sequences to be selected that will distinguish between otherwise closely related genes. This will facilitate selection of oligos and larger regions for expression array. Similarly, a complete and accurate protein sequence will allow proteomics approaches to study gene expression/function Martienssen (2000). For example, complex protein mixtures can be subjected to peptide analysis by mass spectrometry, and a unique matches identified in the genome sequence. This also allows simple comparative analysis to identify protein-coding regions that can then be readily built into gene models. Comparative analysis is far more powerful than compositional analysis for gene finding. Exon-intron structure is typically conserved between species, which should make gene modeling for more accurate. If the right genome is selected, it should be possible to identify conserved regulatory elements in promoters.
Genetrap database: The availability of complete annotated genomic sequence allows each insertion to be annotated automatically by simply searching insertion sites against the genomic sequence for sequence matches at the nucleotide level. Genebank annotation from the nearest gene can then be automatically downloaded and parsed into the Genetrap database on a periodic basis (Tissier et al. 1999). Similarly, three structural proteins (VP6, VP7 and VP4) were sequenced at the DNA level in several rotavirus strains in terms of their recognition and reactively with panels of antibodies (Hoshino and Kapikian, 1994) and their role in virus assembly and infectivity (Patton, 1994). VP6, which makes up inner capsid of rotavirus is the major structural protein of this virus characterized in detail by using mass spectrometry and two-dimensional gel-electrophoresis (Emslie et al., 2000) and to define some post translational modifications. In the characterization of VP6, SA11 had the predicted C-terminus with no modification, and peptide mass fingerprinting data was used to clarify the amino acid sequence at sites where there were conflicts in the DNA sequence (Smith-Huerta et al., 1989).
Feed forward loop (FFL) system: The expression of many genes encoding transcriptional activators in prokaryotes and eukaryotes is up regulated through positive feedback activation. During positive feedback activation, a transcriptional activator binds to its own promoter and thus increases its own expression as well as expression of its target genes. The increased levels of the transcriptional activator can be directly correlated with increased expression of its target genes. Schwechheimer et al. (2000) presented a gene expression system, designated feed forward loop (FFL) system, which makes use of this kind of positive feedback regulation for the expression of plant transgenes. In transiently and stably transformed plant cells, the FFL system yields expression levels of a luciferase reporter gene exceeding the levels observed with the potent CaMV35S promoter. Schwechheimer and his colleagues were able to generate transgenic plants which ubiquitously express high levels of a luciferase reporter gene. The activity of transcription factors is regulated by a variety of mechanisms, e.g. homo-or heterodimerization, phosphorylation, degradation etc.(Schwechheimer and Bevan, 1998). The numerous transcriptional activators vary in their strength and possibly in some cases also in their tissue-specific activity (Schwechheimer et al., 1998). By generating artificial promoters which vary in their responsiveness to a specific transcriptional activator, and by including repressor elements in the promoters, a whole range of promoter/transcription factor combinations of varying strength can be devised and used for FFL gene expression systems: weak FFL systems in which strong promoter activity is required to generate enough transcription factor protein to trigger the autoactivation cycle, and strong FFL systems as suggested by Schwechheimer et al. (2000), that are triggered from weak promoters. However, it can be hypothesized that the presence of high concentrations of transcription factor in the nucleus may be deleterious for eukaryotic cells and that the cells possess mechanisms which allow us to stop expression of these genes. Hence, a regulatory mechanism which excludes the transcription factor from the nucleus, e.g. the glucocorticoid system (Aoyama and Chua, 1997), could be used to overcome the silencing of gene expression in FFL system (Schwechheimer et al. (2000).
Extensive information through cDNA libraries: Targeting expressed genes, through the construction, characterization and the analysis of extensive cDNA libraries, provides information that is potentially of direct relevance to a particular phenotype (Clarke et al., 2000). Due to worldwide efforts in sequencing expressed genes and chromosomal DNA in Arabidopsis and rice, it is now possible to apply the developing knowledge to more complex genomes present in the wheat and barley. This is possible because there is a significant level of conserved gene order and content between the genomes, particularly between wheat and rice (Larik and Soomro, 2000; Larik et al., 2001a, b). An estimate of the total number of genes in rice is approximately 30,000 (Yamamoto and Sasaki, 1997). Panstruga et al. (1998) suggest that although the grass genomes (Poaceae) show great variability in DNA content, there is no evidence that the total number of genes/gene families varies substantially. The concept of homoeology between the genomes and chromosomes of wheat and its relatives implies that similar homoeologies should exist between the individual genes and the polypeptides coded by them (Siddiqui et al., 1972). Of necessity, the homoeology of a given set of polypeptides must be deduced from their degree of chemical relatedness which, ideally should be deduced from their sequence of amino acid polypeptides with similar enzyme properties can be detected by the so called “Zymogram” techniques. The genes for the formation of various iso-enzymes may ultimately be correlated with genes for visible and physiological effects, thus elucidating the biological function of many enzymes (Barber et al., 1968).
Informational Implications of Gene Function in Eukaryotes
The nuclear DNA reversion: Al-saheal and Larik (1985) determined the changes in the gene activity between large (L) and small (S) genotrophs crossed to the PL (plastic) genotypes when grown either at the lower temperature (T2, outside all times) or at the higher temperature (T1, the first 5 weeks in greenhouse). Results demonstrate that (I) the DNA show increased reversion in the crosses, compared with parents at T1 and less reversion in the crosses, than in the parent, at T2; (ii) more reversion occurs in the PL maternal crosses than in the PL paternal crosses; and (iii) the PL genotroph possess a nuclear and a cytoplasmic factor which plays a direct part in determining nuclear DNA changes in the chromosomes of the stable genotypes. However, the difference in DNA is due to a number of repeated DNA sequences and due in part to a difference in the number of rRNA genes, L having about 60% more rRNA DNA than S (Cullis, 1973; Timmis and Ingle, 1973).
Feedback from nucleus to cytoplasm: Information on the genetic control of environmentally induced DNA variation in flax genotrophs (Al-Saheal and Larik, 1987b), revealed that changes occurred in the amount of nuclear DNA of R when the PL nuclear and cytoplasmic factors were introduced by crossing. Tests were made on the plasticity of the F1’s of their reciprocal crosses to PL and R to understand why L and S genotrophs are stable. The results suggest that there may be feedback from nucleus to cytoplasm in Lo, which inactivates the cytoplasmic factor. Possibly a paramutation like mechanism adjusts the different amount of DNA, or heterochromatin between the homologous chromosomes in heterozygotes. L3 and S3 DNA has reverted to same amount as PL. This reversion in DNA appears to be accompanied by an increase in stability rather than in any gain in plasticity.
Plasticity, viewed here as a character determined by nuclear and cytoplasmic factor (Al-Saheal and Larik, 1985b), can be assumed as a regulatory system whose elements, insofar as they are identifiable at the level of the genetic analysis, interact in a manner suggestive of the lactose operon in Escherichia coli (Jacob and Monod, 1961). Changes in the number of repeated sequences seems to be a more likely source of the changes in the amount of nuclear DNA than polyteny. Since a general inducing environment promoting rapid healthy growth (Durrant, 1971) is essential for induction in flax, cell division may be essential for changes in the number of gene sequences to occur. In a specific environment; by increasing the amount of DNA, the number of sequences increases at each cell division and decreases at each cell division in an environment that induces less DNA untill a level is attained where no further changes can occur and the system becomes relatively stable.
Gene activity in coupling and repulsion phases: Heritable changes from H→h and h→H locus in repulsion and coupling phases were studied in flax genotrophs (Al-Saheal and Larik, 1987b). Changes appear to be paramutation-like where one allele alters its homologue on the other chromosome, as a directly or via associated elements. A model is proposed to explain the association between H→h and plant weight. In repulsion, in the large (L) genotroph, H phenotype had changed to the h phenotype at the time of induction by a heterochromatic region extending over this locus. In the small (S) genotroph, which has less DNA, it is assumed that the heterochromatin does not cover the h locus, and so it is a fully active H. The heterochromatic transfer frequencies were higher in coupling than in repulsion. In the heterozygote, stable equilibria of the homozygotes are destroyed and transfer of heterochromatin, or number of reiterated sequences, or a decrease in one homologue and an increase in the other, occur in this region between homologous chromosomes. The amount and direction of the association is dependent upon the frequency of heterochromatic transfer: 0% transfer gives complete positive association; 50% transfer, no association; 100% transfer, complete negative association. This mechanism of heterochromatic transfer preserves the Mendelian ratio of 3:1 of H:h in the F2.
Branching enzymes: A gene encoding wheat endosperm SBE-1 was characterized by Rahman et al. (1999). There are two types of branching enzymes in plants, starch branching enzyme-1 (SBE-1) and starch branching enzyme-II (SBE-11), and both are about 85-90 kda in mass. At the nucleic acid level there is about 65% sequence identity between type I and II, the sequence identity between SBE-1 from different cereals is about 80% overall (Burton et al., 1995; Morell et al., 1995). While SBE-1 and SBE-11 catalyse identical reactions, evidence from mutational and gene expression experiments demonstrate that the enzymes differ in their roles, and biochemical evidence suggests that they differ in their patterns of action (Gaun et al., 1997). In maize (Boyer and Preiss, 1981), rice (Mizuno et al., 1993) and pea (Smith, 1988), null mutation in SBE-11 reduce starch branching and lead to a high amylose phenotype. In contrast, the partial suppression by antisense of SBE-1 activity in the potato tuber leads to subtle alterations in starch physiochemical properties but not to alterations in amylose/amylopectin ratio (Flipse et al., 1996). Mutants lacking SBE-11 activity are not known. Several possible reasons for this can be advanced: (I) SBE-1 may be encoded by multiple genes, (ii) the null mutation does not lead to a phenotype identified in coarse screens for seed morphology or starch structure, or (iii) an SBE-1 mutant is lethal for reasons which are not yet evident. Similarly, three genes encoding the low molecular-weight glutenin sub units (LME-GSs), LMWG-E2 and LMWG-E4, from A-genome diploid wheat species, and LMW-16/10 from a D-genome diploid wheat, were expressed in bacteria by Lee et al. (1999). These proteins were compared with respect to their effects on flour-processing properties such as dough mixing, extensibility and maximum resistance which are the important features in the enduse of wheat food products.
Ectopic expression of genes and aberrant transcripts: Girard and Freeling (1999) have found Lg3-O, a sami dominant neomorphic mutation that transforms regions of leaf blade, auricle and ligule into sheath. The liguleless3 gene is a member of knox class 1 family of homeobox genes and the dominant alleles which define it are due to ectopic expression of the gene in the leaf. They found that the transcripts produced by these alleles are significantly shorter than those of wild type as well as it progenitor, Lg3-O. They used RACE (Rapid Amplification of cDNA Ends) to clone the cDNA corresponding to the Lg3-Or422 transcript and found that the transcripts produced by this allele are being initiated approximately 187 base pairs downstream relative to wild type. Similarly, green-fluorescent compounds induced by ectopic expression of P gene in maize was studied by Lin et al. (2000). The maize P gene, R2R3 myb transcription factor, controls 3-deoxy flavonoid and phlobaphene biosynthesis. In the pericarp, P regulates the accumulation of a subset of flavonoid biosynthetic genes (C2, Chi1 and A1). The ectopic expression of P in cultured BMS cells induces the accumulation of distinct classes of flavonoid and phenylpropanoid compounds, as well as orange-fluorescent bodies.
The P1 gene encodes a transcriptional regulator of red phlobaphene pigment biosynthesis. A P1-rr allele conditions uniform red pigmentation of the pericarp, cob glume, husks, silks and tassel glumes, whereas plants carrying a P1-wr allele lack pericarp pigmentation, but have uniform, darkred cob glumes and pigmented margins on the husks and tassel glumes. When maize plants were transformed with constructs containing the P1-rr promoter deriving either P1-rr or P1-wr cDNA sequences, the transgenes promoted pigmentation in the floral organs as well as in the vegetative organs of plants. This pattern of transgene expression is comparable to that caused by Ufo1 (unstable factor for orange), a dominant allele that induces phlobaphene production in vegetative organs when combined with a P1-wr allele (Cocciolone et al., 2000).
Concluding remarks: In present review we have critically examined and discussed the gene structure and function in prokaryotes and eukaryotes with particular reference to DNA information technology. Methods of experimentation used to obtain information for bioinformatic analysis include electrophoresis, chromatography and a relatively new area biochips, which are miniaturized devices that can make biological experimentation more efficient. Functionally, biochips include DNA chips, protein chips and lab chips. Biochips contain either immobilized DNA strands (DNA chips), immobilized protein strands (protein chips) are interconnected channels with fluid propulsion and control systems etched into glass, silicon, quartz or plastic (lab chips) that respectively permit gene, protein and expression system analysis on a single chip. Biochips are being increasingly used to accelerate drug discovery and development. From a modest start of $ 12 million in 1997 the biochip market is expected to balloon $ 632 million in 2005.
REFERENCES
- Al-Saheal, Y.A. and A.S. Larik, 1985. Genetic parameters of flax genotrophs. V. Behaviour of nuclear DNA in parental and F1 generations. Can. J. Genet. Cytol., 27: 6-11.
Direct Link - Al-Saheal, Y.A. and A.S. Larik, 1987. Linkage studies of the h-gene with plant weight in flax genotrophs. Theor. Applied Genet., 73: 343-349.
CrossRefDirect Link - Aoyama, T. and N.H. Chua, 1997. A glucocorticoid-mediated transcriptional induction system in transgenic plants. Plant J., 11: 605-612.
PubMedDirect Link - Barber, N.N., C.J. Driscoll, P.M. Long and R.S. Vickery, 1968. Protein genetics of wheat and homoeologous relationships of chromosomes. Nature, 281: 450-452.
CrossRefDirect Link - Beadle, G.W. and E.L. Tatum, 1941. Genetic control of biochemical reactions in Neurospora. Proc. Natl. Acad. Sci., 27: 499-506.
Direct Link - Boyer, C.D. and J. Preiss, 1981. Evidence for independent genetic control of the multiple forms of maize endosperm branching enzymes and starch synthases. Plant Physiol., 67: 1141-1145.
PubMedDirect Link - Burton, R.A., J.D. Bewley, A.M. Smith, M.K. Bhattacharya and H. Tatga et al., 1995. Starch branching enzymes belonging to distinct enzyme families are differentially expressed during pea embryo development. Plant J., 7: 3-15.
PubMedDirect Link - Chargaff, E., 1950. Chemical specificity of nucleic acids and mechanism of their enzymic degradation. Experentia, 6: 201-209.
CrossRefDirect Link - Clarke, B.C., M. Hobbs, D. Skylas and R. Appels, 2000. Genes active in developing wheat endosperm. Funct. Integr. Genomics, 1: 44-55.
Direct Link - Durrant, A., 1971. The induction and growth of flax genotrophs. Heredity, 27: 277-298.
CrossRefDirect Link - Emslie, K.R., M.P. Molloy, C.R.M. Bararadi, J. Jardine, M.R. Wilkins, A.R. Bellamy and K.L. Williams, 2000. Serotype classification and characterization of the rotavirus SA11 VP6 protein using mass spectrometry and two dimentional gel electrophoresis. Funct. Integr. Genomics, 1: 12-24.
PubMedDirect Link - Fincham, J.R.S., 1959. The biochemistry of genetic factors. Ann. Rev. Biochem., 28: 343-364.
CrossRefDirect Link - Fraenkel-Conrat, H., 1956. The role of nucleic acid in the reconstitution of active tobacco mosaic virus. J. Am. Chem. Soc., 78: 882-883.
CrossRefDirect Link - Gaun, H.P., P. Li, J. Imparl-Radosevich, J. Priess and P. Keeling, 1997. Comparing the properties of Escherichia coli branching enzyme and maize branching enzyme. Arch Biochem. Biophys., 342: 92-98.
CrossRefDirect Link - Gierer, A. and G. Schramm, 1956. Infectivity of ribonucleic acid from tobacco mosaic virus. Nature, 177: 702-703.
CrossRefDirect Link - Girard, L. and M. Freeling, 1999. Mutator-suppressible liguleless3 alleles produce altered transcripts. Maize Genet. Coop. Newslett., 73: 14-14.
Direct Link - Jacob, F. and J. Monod, 1961. Genetic regulatory mechanisms in the sysnthesis of protein. J. Mol. Biol., 3: 318-356.
PubMedDirect Link - Larik, A.S., K.A. Siddiqui and Z.A. Soomro, 2001. Significant contributions of innovative cytogenetics to contemporary plant improvement. Proc. Pak. Acad. Sci., 38: 59-74.
Direct Link - Larik, A.S., K.A. Siddiqui, Z.A. Soomro and F. Khowaja, 2001. Genome evolution in eukaryotes-a futuristic vision. Proc. Pak. Acad. Sci., 38: 181-193.
Direct Link - Lee, Y.K., F. Bekes, R. Gupta, R. Appels and M.K. Morell, 1999. The low-molecular-weight glutenin subunit proteins of primitive wheats. I. Variation in A-genome species. Theor. Applied Genet., 98: 119-125.
CrossRefDirect Link - Martienssen, R.A., 2000. Weedingout the genes: The Arabidopsis genome project. Funct. Integr. Genomics, 1: 2-11.
CrossRefDirect Link - McConkey, E.H. and J.W. Hopkins, 1964. The relationship of the nucleous to the synthesis of ribosomal RNA in Hela cells. Proc. Natl. Acad. Sci. USA., 51: 1197-1204.
Direct Link - Mizuno, K., T. Kawasaki, H. Shimada, H. Satoh and E. Kobayashi et al., 1993. Alteration of the structural properties of starch components by the lack of an isoform of starch branching enzyme in rice seeds. J. Biol. Chem., 268: 19084-19091.
Direct Link - Morell, M.K., S. Rahman, S.L. Abrahams and R. Appels, 1995. The biochemistry and molecular biology of starch synthesis in cereals. Aust. J. Plant Physiol., 22: 647-660.
CrossRefDirect Link - Panstruga, R., R. Buschges, P. Riffanelli and P. Schulze-lefert, 1998. A contiguous 60 kb genome stretch from barley reveals molecular evidence for gene islands in a monocot genome. Nucleic Acids Res., 26: 1056-1062.
PubMedDirect Link - Perry, R.P. 1964. Role of Nucleolus in ribonucleic acid metabolism and other cellular processes. Natl. Cancer Inst. Monogr., 14: 73-89.
PubMedDirect Link - Rich, A., A. Nordheim and A.H.J. Wang, 1984. The chemistry and biology of left handed zDNA. Ann. Rev. Biochem., 53: 791-846.
CrossRef - Schwartz, D., 1965. Genetic control of the pH 7.5 Esterase in maize. Natl. Cancer Inst. Monogr., 18: 9-14.
PubMedDirect Link - Schwechheimer, C., C. Smith and M. Bevan, 1998. The activities of acidic and glutamine-rich transcriptional activation domains in plant cells: Design of modular transcription factors for high level expression. Plant Mol. Biol., 36: 195-204.
PubMedDirect Link - Schwechheimer, C., F.M.K. Corke, C.H. Smith and M. Bevan, 2000. Transactivation of a target gene through feedforward loop activation in plants. Funct. Integr. Genomics, 1: 35-43.
CrossRefDirect Link - Smith, A.M., 1988. Major differences in isoforms of starch branching enzyme between developing embryos of round and wrinkled seeded peas (Pisum sativum L.). Planta, 175: 270-279.
CrossRefDirect Link - Timmis, J.N. and J. Ingle, 1973. Environmentally induced changes in rRNA gene redundancy. Nat. New Biol., 244: 235-236.
PubMedDirect Link - Tissier, A.F., S. Marillonnet, V. Klimyuk, K. Patel, M.A. Torres, G. Murphy and J.D. Jones, 1999. Multiple independent defective suppressorCmutator transposon insertions in azabidopsis. A tool for functional genomics. Plant Cell, 11: 1841-1852.
Direct Link - Watson, J.D. and F.H.C. Crick, 1953. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature, 171: 737-738.
Direct Link - Watson, J.D. and F.C. Crick, 1953. Genetical implications of the structure of deoxyribose nucleic acid. Nature, 171: 964-967.
CrossRefDirect Link - Yamamoto, K. and T. Sasaki, 1997. Large scale EST sequencing in rice. Plant Mol. Biol., 35: 135-144.
PubMedDirect Link