
S. Farid Uddin Farhad
Industrial Physics Division, BCSIR Laboratories, Dhaka-1205, Bangladesh
K.M. Abedin
Department of Physics, University of Dhaka, Dhaka-1000, Bangladesh
M. Rafiqul Islam
Department of Physics, University of Dhaka, Dhaka-1000, Bangladesh
Aminul I. Talukder
Department of Physics, University of Dhaka, Dhaka-1000, Bangladesh
A.F.M.Y. Haider
Department of Physics, University of Dhaka, Dhaka-1000, Bangladesh
Journal of Applied Sciences
Year: 2009 | Volume: 9 | Issue: 8 | Page No.: 1538-1543







ABSTRACT
In this study, a homemade laser Raman spectrometer was used to examine the Raman spectra of different edible oils, such as canola, corn, soybean, sunflower, palm and coconut oils. To avoid severe fluorescence effects which tend to mask the weak Raman lines, Raman bands in the 2800-3200 cm-1 region resulting from unsaturated and saturated carbon bonds in the fatty acids were chosen and measured the TUSFA/TFA (total unsaturated fatty acid/total fatty acid content) ratios of the various samples. Good correlation of the ratios of TUSFA/TFA of different edible oils with that calculated from manufacturers data was obtained, enabling us to measure unsaturation/total fatty acids ratio quickly and easily for any unknown oils and hence the measure the degree of possible adulteration.
PDF Abstract XML References Citation
How to cite this article
S. Farid Uddin Farhad, K.M. Abedin, M. Rafiqul Islam, Aminul I. Talukder and A.F.M.Y. Haider, 2009. Determination of Ratio of Unsaturated to Total Fatty Acids in Edible Oils by Laser Raman Spectroscopy. Journal of Applied Sciences, 9: 1538-1543.
DOI: 10.3923/jas.2009.1538.1543
URL: https://scialert.net/abstract/?doi=jas.2009.1538.1543
DOI: 10.3923/jas.2009.1538.1543
URL: https://scialert.net/abstract/?doi=jas.2009.1538.1543
INTRODUCTION
Raman spectroscopy has become a very important tool for chemical analysis in many industrial and commercial products (Sadeghi-Jorabchi et al., 1991; Baeten et al., 1996; Glen and Horvat, 1972; Baeten et al., 1998; Baeten and Aparicio, 2000; Barbara et al., 2005). Since molecules emit their characteristic Raman spectra, the identification and analysis of a chemical species can be performed very rapidly by observing their Raman spectra. If the sample is a complex mixture of chemical species, the different constituents can be identified and quantified by measuring the intensities of their respective Raman lines or bands, since these intensities can be correlated with the abundances in the samples. The method requires no chemical reagents and can be performed very quickly in the laboratory or on the field. Analysis of edible oils have been performed using the methods described elsewhere (Baeten et al., 1996; Liliana et al., 2006; Morton and Barbara, 1967; Sacco et al., 2000; Marcos et al., 2002) and the detection of adulteration has also been accomplished. Usually characteristic Raman bands in the low frequency region is used as the fingerprint region (Sadeghi-Jorabchi et al., 1991; Baeten et al., 1996; Glen and Horvat, 1972) for this type of analysis, since these are the easiest to observe and measure and are directly related to the structure of the molecules.
Edible oils usually contain significant amounts of fluorescent material such as carotenes, which tends to mask the weak Raman lines (Abedin et al., 2008; Chase, 1987; Parker et al., 1999) especially if the Raman spectra is excited by lasers such as Argon ion or He-Ne lasers. The alternative is to use an FT-Raman system, with excitation at 1064 nm (Chase, 1987), but such systems are complicated and expensive. Another alternative is to use a laser source with energy lower than the excited level resulting in the fluorescence.
A simple but powerful laser Raman system was constructed using a dielectric interference filter in our laboratory to acquire Raman spectra of different samples (Abedin et al., 2008). In this study, we used the Raman system to measure Raman spectra of a number of edible oils and subsequent analysis is performed from the measured spectra, with an emphasis in the high-frequency 2800-3200 cm-1 region, enabling us to measure and quantify the unsaturation in edible oils.
MATERIALS AND METHODS
Instrument setup: The schematic diagram of the laser Raman system is shown in Fig. 1. Monochromatic light (at λ = 632.8 nm) from a 20 mW unpolarized helium-neon laser (Uniphase) is incident by a short-focus (f = 50 mm) lens L1 on the liquid sample kept in a suitable sample holder (quartz or borosilicate glass bottles).
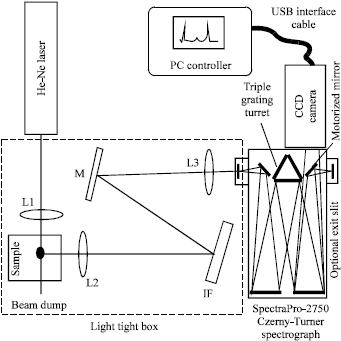 | |
| Fig. 1: | Schematic diagram of the constructed Raman system. IF: Interference filter; L1 Expanding lens, L2: Collection lens, L3: Focusing lens, M: Mirror |
Light scattered at an angle of 90° to the incident laser beam was collected by another short-focal length lens L2 (f = 50 mm, d = 50 mm) and collimated. The light, which contains the Rayleigh as well as the Raman scattered light, is incident on the 3-cavity dielectric interference filter IF (diameter 25 mm, Coherent). The filter has a peak transmission of 80% at 632.8 nm and a FWHM of about 6 nm. As a result of multiple interference inside the filter, light in a narrow band of wavelengths near 632.8 nm (Rayleigh-scattered light) is mostly transmitted through it, while light outside this band is mostly reflected. The reflected light, containing the Raman-scattered energy, is directed by a mirror (M) towards the spectrograph input. The light is focused on the input slit of the spectrograph by the lens L3 (f = 75 mm). The entire optics on the input side is kept inside a light-tight box in order to reduce the effect of stray light which can interfere with the sensitive Raman measurements. The box has a door on one side and by opening it sample vial can be placed in the system for examination.
The spectrograph (Acton 2750SP) is a Czerny-Turner system using an interchangeable 3-grating turret and has a focal length of 750 mm. The resolution of the system for a 300 lines mm-1 diffraction grating and 10 μ slit width was about 0.1 nm (about 2.5 cm-1). Each selected grating can be scanned by computer control. The input side has an adjustable slit but the output side has none. The light falls on a deep-cooled CCD camera (Princeton Instruments PIXIS 100 B) attached to the output port. The camera has a 1340x100 pixel CCD chip cooled to -75 °C by Peltier coolers in order to reduce dark noise. The acquired data (spectrum) can be transferred to the PC by a USB cable and all camera functions, such as setting of acquisition time, camera gain, cosmic ray elimination, as a well as grating scanning can be performed by WinSpec/32 software installed in the attached PC running under Windows XP operating system. The software can also automatically perform subtraction of background noise from the signal, as well as gluing spectra from two adjacent, overlapping spectral regions to make one spectrum. The spectrum can be directly shown on the PC monitor with the abscissa either in nm, in wave numbers (cm-1) or in relative Raman shifts in wavenumbers.
RESULTS AND DISCUSSION
Acquisition of Raman spectrum in the 2800-3200 cm-1 region: Raman spectra of edible oils, like soybean, sunflower, canola, palm oil, corn oil, coconut oil etc., were acquired. These oils are collected in sealed containers from supermarket in order to ensure the authenticity of the samples. In some oils, we observed very strong broadband fluorescence in the stokes Raman region. For example, in soybean oil, very strong fluorescence is emitted in such quantities that it completely masks the Raman signal (Fig. 2) when using the He-Ne laser as the excitation source. Almost all the Raman signatures are fully concealed by the fluorescence signal emitted by the carotenes. Due to this difficulty, we scanned the spectrometer to the long-wavelength side and observed the Raman lines (relative Raman shift) in the 2800 to 3200 cm-1 region. We acquired the spectra and found that it contained usable information.
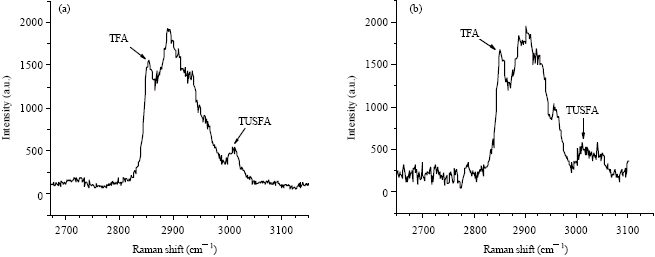
Data analysis: A fourth order polynomial line to the fluorescence background was fitted (best fit) and was subtracted from the total spectrum. The Raman lines in the 2800 to 3200 cm-1 region clearly stood out after this subtraction process. Figure 3 shows the subtracted spectrum in this frequency region for soybean oil. The spectra for other oils such as canola, sunflower, corn, palm and coconut oil are shown in Fig. 4a-e. In all these cases a five point adjacent averaging method was used to smoothen out the curve.
Three prominent features can be identified in this region. The shoulder band at 2855 cm-1, the strong band at 2970 cm-1 and the band at 3105 cm-1. The first band at 2855 cm-1 is due to symmetric C-H stretching vibration of -CH2-groups, the intensity of which is a measure of the Total Fatty Acid (TFA) chain of the oil molecules (Baeten et al., 1996).
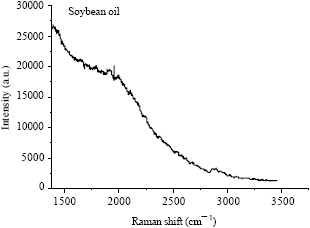 | |
| Fig. 2: | Raman spectrum of soybean oil masked by strong fluorescence |
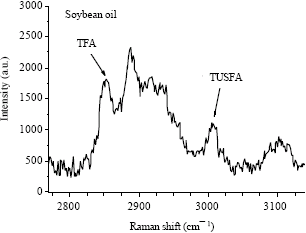 | |
| Fig. 3: | Raman spectrum of soybean oil after polynomial fitted background subtraction and five point adjacent averaging |
The second band at 2970 cm-1 is attributed to the C-H stretching vibration of methyl groups. The third band at 3105 cm-1 is due to C-H symmetric stretching vibrations of =C-H groups, the intensity of which is a measure of the total unsaturated fatty acid (TUSFA) chain ( Baeten et al., 1996). The first and third vibrational modes attributable to TFA and TUSFA are shown in Fig. 5 (Colthup et al., 1975; Sadeghi-Jorabchi et al., 1991).
The different vibrational modes of the fatty acid chain of the oil molecules which are of interest in the Raman analysis of edible oils (Baeten et al., 1998) are shown in Table 1. The ratios of the peaks of the bands at 3015 and 2855 cm-1 were calculated for five Raman spectra for each of the edible oils and the average was taken as the representative value of TUSFA/TFA content for that particular oil. The ratio of the peaks of the TUSFA and TFA lines could be taken as a rough estimate of the ratio of their intensities.
| Table 1: | Major Raman scattered bands typically used in Edible oil analysis |
 | |
| Baeten et al. (1998) | |
| Table 2: | The TUSFA/TFA ratios measured by Raman spectroscopy and calculated from the manufacturers’ data |
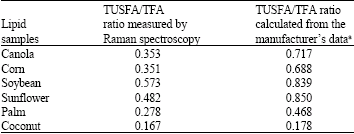 | |
| aRatios calculated from the unsaturated and total fatty acid contents as provided by the manufacturer (claimed to be measured by gas chromatography) | |
We called this ratio TUSFA/TFA ratio (measured by Raman spectroscopy) and this is shown in Table 2, which also shows the TUSFA/TFA ratio calculated from data provided by the manufacturers of the oils. The correlation between our results and that calculated from the manufacturers’ data is shown in Fig. 6. A good degree of correlation (R2~0.92) between the ratios measured by the present method using Raman spectroscopy and that calculated from manufacturers data is seen. Despite of an excellent correlation between the results of Raman spectroscopy and manufacturers’ data, the TUSFA/TFA ratio was observed to be consistently lower in the present study. This may be attributed to the following facts. In case of TFA line, the peak could have been over estimated because of the contribution of the leading edge of the Raman shift line at 2970 cm-1due to the C-H stretching vibrational mode of methyl groups. But the TUSFA line was better resolved and there was no such effect. Therefore in the estimated ratio of TUSFA to TFA, the peak of TFA line could have been over estimated consistently in all cases. Hence the TUSFA to TFA ratio was systematically observed to be lower as compared with manufacturer’s data.
In conventional Raman spectroscopy of edible oils, it is customary to use low frequency Raman bands in the so called fingerprint region (800-1800 cm-1) to characterize the unsaturation level of the fatty acid chain (Sadeghi-Jorabchi et al., 1991; Glen and Horvat, 1972; Baeten et al., 1998; Baeten and Aparicio, 2000; Barbara et al., 2005).
 | |
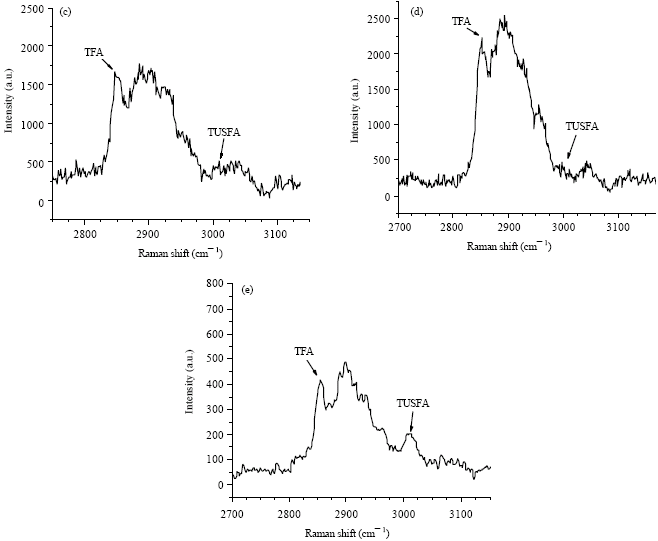 | |
| Fig. 4: | Raman spectra of edible oils after background subtraction and five point adjacent averaging, (a) canola oil, (b) corn oil, (c) palm oil, (d) coconut oil and (e) sunflower oil |
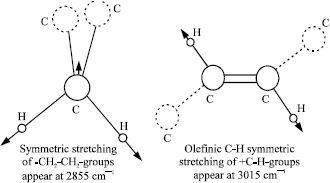 | |
| Fig. 5: | Vibrational modes of the key bands in the high frequency region |
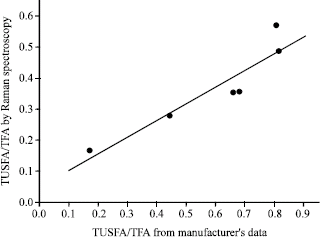 | |
| Fig. 6: | Correlation between the TUSFA/TFA ratio measured by the present method using Raman spectroscopy and that calculated from data provided by the manufacturer |
However, if very strong fluorescence light is emitted from the sample, it is impossible to measure the relatively weak Raman features riding on the fluorescence signal, particularly if visible lasers are used for excitation. To overcome this difficulty, we selected Raman bands in the high frequency region (2800 to 3200 cm-1) as stated earlier, which are usually not used for this type of analysis. These high frequency bands are easily observable even in the presence of very strong fluorescence. This method sets a standard for TUSFA/TFA ratio for pure edible oils. For example, we have determined the TUSFA/TFA ratio for pure canola oil as 0.353. For any unknown samples of Canola oil if this ratio is significantly different from 0.353, there must be every reason to suspect adulteration in that sample of canola. So, this experimental method, which uses the high frequency Raman bands, can be used for determination of adulteration in edible oils. A good degree of correlation between the TUSFA/TFA ratio measured by using this high frequency Raman bands and that calculated from the data provided by the manufacturer indicates that this method using the high frequency bands can be used for this type of analysis.
The degree of unsaturation is very important for the quality of fats and oils since unsaturated bonds are considered to have beneficial health effects (Barbara et al., 2005). Low quality edible oils have higher saturated fatty acid contents, which may be added as an adulterant to good quality edible oil. Therefore, a measure of the ratio of TUSFA/TFA can be used as indicator of adulteration by low quality oil. Chemical methods such as Gas Chromatography (GC), High Performance Liquid Chromatography (HPLC) etc. are available to measure the unsaturation level, but these are time consuming and require the use of expensive reagents. Compared to these methods, Raman spectroscopy is considered to be very convenient, inexpensive, rapid and non-destructive method, suitable for even on-line analysis.
In conclusion, we have shown in this study that it is possible to measure the level of unsaturation using the very simple dispersive laser Raman system employing an inexpensive He-Ne laser for excitation and a dielectric interference filter for corresponding Rayleigh filtering. In the present case, the difficulty arising out of fluorescence emission due to the use of He-Ne laser for excitation, is overcome by observing Raman bands at high frequency region, a band which is not conventionally used for measuring the unsaturation level. It is found that good correlation exists between the TUSFA/TFA ratio measured from these Raman bands and the TUSFA/TFA ratio measured by chemical methods. Therefore, the present method could be used for reliable and effective markers for the measurement of unsaturation level.
ACKNOWLEDGMENT
This study was done in the Nonlinear Optics and Laser Spectroscopy Laboratory of the Center of Advanced Research in Physical, Chemical, Biological and Pharmaceutical Sciences (Center of Excellence) of Dhaka University.
REFERENCES
- Abedin, K.M., S.F.U. Farhad, M.R. Islam, A.I. Talukder and A.F.M.Y. Haider, 2008. Construction and operation of a dispersive laser Raman spectrograph using interference filter. J. Bangladesh Acad. Sci., 32: 121-129.
CrossRefDirect Link - Baeten, V., M. Meurens and M.T. Morales-R.Aparicio, 1996. Detection of virgin olive oil adulteration by FT Raman spectroscopy. J. Agric. Food Chem., 44: 2225-2230.
CrossRefPubMedDirect Link - Baeten, V., P. Hourant, M.T. Morales and R. Aparicio, 1998. Oil and fat classification by FT-Raman spectroscopy. J. Agric. Food Chem., 46: 2638-2646.
Direct Link - Baeten, V. and R. Aparicio, 2000. Edible oils and fats authentication by FT-Raman spectroscopy. Biotechnol. Agron. Soc. Environ., 4: 196-203.
Direct Link - Barbara, M., B. Lendl, A. Molina-Diaz and M.J. Ayora, 2005. Direct monitoring of lipid oxidation in edible oils by FT-Raman spectroscopy. Chem. Phys. Lipids, 134: 173-182.
CrossRefDirect Link - Chase, B., 1987. Fourier transform Raman spectroscopy. Mikrochim. Acta, 93: 81-91.
CrossRefDirect Link - Glen, F.B. and R.J. Horvat, 1972. Raman spectroscopic analysis of the cis/trans isomer composition of edible vegetable oils. J. Am. Oil Chem. Soc., 49: 494-494.
CrossRefDirect Link - Giacomelli, L.M., M. Mattea and C.D. Ceballos, 2006. Analysis and characterization of edible oils by chemometric methods. J. Am. Oil Chem. Soc., 83: 303-308.
CrossRefDirect Link - Marcos, L.I., J.L. Perez Pavon, M.E. Fernandez Laespada, C. Garcia Pinto and B. Moreno Cordero, 2002. Detection of adulterants in olive oil by headspace- mass spectrometry. J. Chromatogr., 945: 221-230.
PubMed - Morton, B. and A.B. Barbara, 1967. Rapid determination of olefin position in organic compounds in microgram range by ozonolysis and gas chromatography. Ana. Chem., 39: 1131-1135.
CrossRef - Parker, S.F., S.M. Tavender, N.M. Dixon, H. Herman, K.P.J. Williams and W.F. Maddams, 1999. Raman spectrum of β-carotene using laser lines from green (514.5nm) to near-infrared (1064nm): Implications for the characterization of conjugated polyenes. Applied Spectros., 53: 86-91.
CrossRefDirect Link - Sadeghi-Jorabchi, H., R.H. Wilson, P.S. Belton, J.D. Edwards-Webb and D.T. Coxon, 1991. Quantitative analysis of oils and fats by FT Raman spectroscopy. Spectrochim. Acta., 47: 1449-1458.
CrossRefDirect Link - Sacco, A., M.A. Brescia, V. Liuzzi, F. Renjero, C. Guillou, S. Ghelli and P. Van der Meer, 2000. Characterization of Italian olive oils based on analytical and nuclear magnetic resonance determinations. J. Am. Oil Chem. Soc., 77: 619-625.
CrossRefDirect Link