
G. Pournima
Department of Oral and Maxillofacial Pathology, Terna Dental College and Hospital, Navi Mumbai Nerul, Maharashtra State, India
Y. Monica
Department of Oral and Maxillofacial Pathology, Terna Dental College and Hospital, Navi Mumbai Nerul, Maharashtra State, India
S. Meghna
Department of Oral and Maxillofacial Pathology, Terna Dental College and Hospital, Navi Mumbai Nerul, Maharashtra State, India
European Journal of Dentistry and Medicine
Year: 2012 | Volume: 4 | Issue: 1 | Page No.: 8-13







ABSTRACT
Craniosynostosis are heterogenous group of syndromes characterized by premature sutural fusion that occurs individually or relating to other anamolies. The type of the obliterated sutures determines the type of craniosteinosis. There are over 150 syndromes associated with it. Crouzon syndrome is an example of such a syndrome caused by premature obliteration and ossification of two or more sutures, most commonly coronal and sagittal. Described by a French neurosurgeon in 1912, it is a rare genetic disorder. Crouzon syndrome is caused by mutation in the Fibroblast Growth Factor Receptor 2 (FGFR2) gene. This disorder is characterized by distinctive malformations of skull and face (craniofacial region). Premature cranial suture closure is the most common skull abnormality. The case of a 7-year-old Indian boy with Crouzon Syndrome which is one of syndromes associated with synostosis, is presented. He presented with a cranial deformity, maxillary hypoplasia and exophthalmos. The clinical, characteristic radiological features and investigations carried out, along with treatment of this patient are discussed as part of multidisciplinary management.
PDF Abstract XML References Citation
Received: March 14, 2011;
Accepted: April 19, 2011;
Published: March 20, 2012
How to cite this article
G. Pournima, Y. Monica and S. Meghna, 2012. Crouzon Syndrome: A Case Report. European Journal of Dentistry and Medicine, 4: 8-13.
DOI: 10.3923/ejdm.2012.8.13
URL: https://scialert.net/abstract/?doi=ejdm.2012.8.13
DOI: 10.3923/ejdm.2012.8.13
URL: https://scialert.net/abstract/?doi=ejdm.2012.8.13
INTRODUCTION
Crouzon syndrome is a rare genetic disorder characterized by premature closure of cranial sutures, exophthalmos and mid facial hypoplasia. Crouzon syndrome occurs in approximately 1 in 25,000 births world wide and 16.5 per 1,000,000 live births in United States. It makes up approximately 4.8% of all cases of, craniosynostosis, making it the most common syndrome within the craniosynostosis group. It may be transmitted as an autosomal dominant genetic condition or appear as a mutation .No known race or sex predilection exists (Cohen and Kreiborg, 1992). The majority of the patients with Crouzon syndrome have mutations in the extracellular immunoglobulin III domain of the Fibroblast Growth Receptors 2(FGFR2)gene (Jabs et al., 1994). The differential diagnosis of Crouzon syndrome includes Apert syndrome, Pfeiffer, Jackson-Weiss, Carpenter and Saethre-Chotzen syndrome. Crouzon syndrome is distinguishable from other craniosynostosis syndromes by lack of hand and/or foot abnormalities (Bowling and Burstein, 2006). Multiple staged surgeries are the general treatment plan for patients with Crouzon syndrome (Posnick and Ruiz, 2000). In this article, we present a case of Crouzon syndrome in a boy aged 7 years.
CASE REPORT
A 7 year old boy who had come for routine dental check up, was referred to the Dept of Oral Pathology for evaluation of dysmorphic features. On general examination the boy was of a short stature, had wide nasal bridge (hypertelorism) and mild exophthalmos (Fig. 1). The boy also complained of recurrent headaches and transient visual obscurations. The patient did not have any digital abnormalities or hearing deficit. Intraoral examination revealed high arched palate, hypo plastic maxilla leading to pseudo prognathism of mandible and class III malocclusion. No dental aplasia was present. On inquiring the mother reported that she had delivered when she was 35 years old. History revealed that these features started developing since he was a child and the severity gradually increased over a period of time. There was no significant positive family history. The patient was evaluated by an Opthalmologist in view of visual complaints which revealed papilloedema suggestive of raised intracranial pressure. Taking into consideration the above mentioned findings, following investigations were done.
INVESTIGATIONS
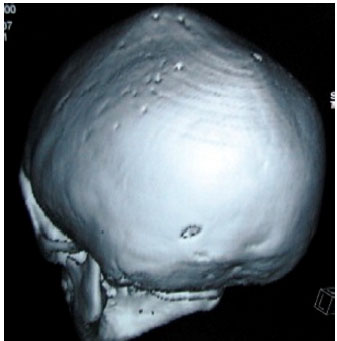
3D Volume rendered images of the skull showed fusion of the coronal and the sagittal sutures, prominent convolutional markings suggestive of copper beaten appearance, shallow orbits and widened intercanthal distance (Fig. 2, 3, 5).
CT scan Brain showed prominent convolutions in the inner table of the skull (Fig. 4).
The overall radiological features confirmed the diagnosis of Crouzon syndrome.
The case was referred to:
| • | Neurosurgery department in view of the raised intracranial pressure and craniosynostosis and the procedure of suturectomy with cranial morcellation was advised |
| • | Orthodontic dept for correction of malocclusion |
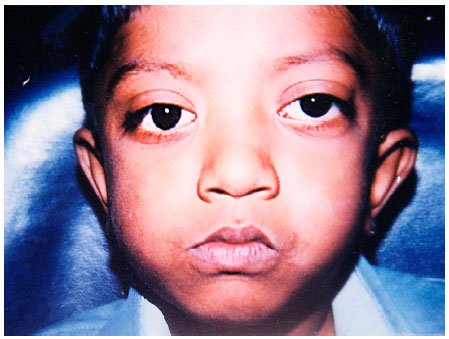 | |
| Fig. 1: | Shows mild exophthalmos and hypertelorism |
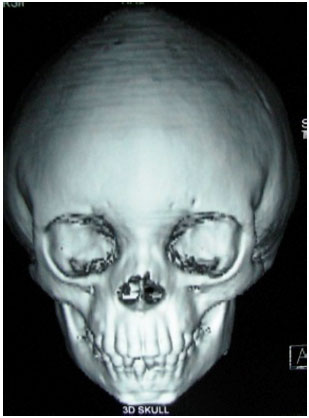 | |
| Fig. 2: | 3D Skull showing fusion of the coronal sutures |
 | |
| Fig. 3: | 3D Skull showing fusion of the sagittal and the coronal sutures |
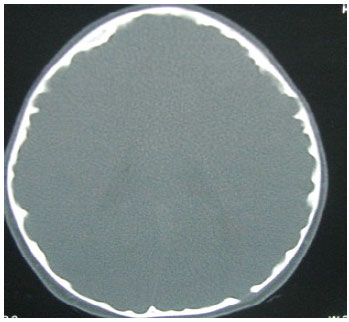 | |
| Fig. 4: | CT scan demonstrating convolutions on the inner calvarium of the Skull |
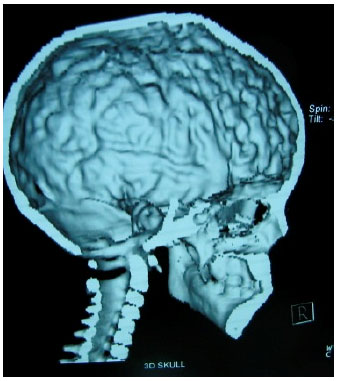 | |
| Fig. 5: | 3D Skull showing a copper beaten appearance |
DISCUSSION
Octave Crouzon (1874-1938), a French neurologist, in 1912, described the hereditary syndrome of craniofacial synostosis in a mother and son. He described the triad as skull deformities, facial anamolies and exophthalmos now known as Crouzon syndrome. It is an autosomal dominant disorder with complete penetrance and variable expressivity (Jabs et al., 1994; Bowling and Burstein, 2006).
The cause is attributable to mutations in the fibroblast growth factor receptor-2 (FGFR2)gene which is mapped to chromosome locus 10q25-10q26,but exhibits locus heterogeneity with causal mutations in FGFR-2 (Crouzon syndrome) and FGFR3 (Crouzon syndrome with Acanthosis nigricans) in different affected individuals (Jabs et al., 1994).
The most common ocular abnormalities are shallow orbits, ocular proptosis, orbital hypertelorism, strabismus, papilloedema, optic atropy, exoposure keratitis and visual loss. There also have been rare occurances of nystagmus, iris coloboma, aniridia, anisocoria, microcornea, megalocornea, cataract, ectopia lentis, blue sclera, glaucoma and luxation of the eye (Bowling and Burstein, 2006).
Headaches and seizures are attributable to elevated intracranial pressure. Conductive hearing loss is common owing to ear canal steinosis or atresia. Upper airway obstruction develops secondary to septal deviation, mid nasal abnormalities, choanal abnormalities and nasopharyngeal narrowing and can lead to acute respiratory distress. Acanthosis nigricans is the main dermatological manifestation in 5% cases of Crouzon syndrome (Cohen, 1975).
In the mouth, short upper lip, hypoplastic maxilla, relative mandibular prognathism, malocclusions and v shaped maxillary dental arch have been reported, as has narrow, high, or cleft palate and bifid uvula (Chen, 2004).
Apert syndrome, Pfeiffer syndrome, Carpenter syndrome, Saethre-Chotzen syndrome are the commonest differential diagnosis to Crouzon syndrome (Bowling and Burstein, 2006).
Radiographs, Magnetic Resonance Imaging (MRI) scans, genetic testing, X-rays and CT scans can be used to confirm the diagnosis. Ultrasonic prenatal diagnosis of Crouzon syndrome has been reported, however molecular testing is more accurate and reliable than ultrasonography. C.T scan brain shows signs of raised intracranial pressure, fusion of coronal and sagittal sutures and 3D images will reveal copper beaten appearance.
Preimplantation genetic diagnosis for Crouzon syndrome by blastomere biopsy samples from cleavage-stage embryos may be detected by mutational analysis (Abou-Sleiman et al., 2002).
The treatment is multidisciplinary and multiple staged surgeries are recommended (Posnick and Ruiz, 2000). .Early craniectomy with frontal bone advancement is most often indicated to prevent or treat increased intracranial pressure. If necessary, midfacial advancement and jaw surgery can be done to provide adequate orbital volume, reduce the exophthalmos and correct the occlusion to appropriate functional position. Prognosis depends on the severity of malformations. Innovations in craniofacial surgery have enabled patients to achieve their full potential by maximizing their opportunities for intellectual growth, physical competence and social interaction.
CONCLUSION
Crouzon syndrome should be managed as early as possible as it results in poor cosmetic appearance and results in other complications like mental retardation, airway obstruction and decreased visual acuity as the age advances. With proper treatment, these patients can be productive and active members of the main stream society.
REFERENCES
- Abou-Sleiman, P.M., A. Apessos, J.C. Harper, P. Serha and J.D.A. Delhanty, 2002. Pregnancy following preimplantation genetic diagnosis for Crouzon syndrome. Mol. Hum. Reprod., 8: 304-309.
CrossRef - Cohen, Jr. M.M., 1975. An etiologic and nosologic overview of craniosynostosis syndromes. Birth Defects Orig. Artic. Ser., 11: 137-184.
PubMed - Cohen, M.M. Jr. and S. Kreiborg, 1992. Birth prevalence studies of the Crouzon syndrome: Comparison of direct and indirect methods. Clin. Genet., 41: 12-15.
CrossRef - Posnick, J.C. and R.L. Ruiz, 2000. The craniofacial dysostosis syndromes: Current surgical thinking and future directions. Cleft Palate Craniofac. J., 37: 433-433.
PubMed