
Arun M. Xavier
Department of Pediatric Dentistry, Amrita School of Dentistry, Cochin, India
Kavita Rai
Department of Pedodontics and Preventive Children Dentistry, A.B. Shetty Memorial Institute of Dental Sciences, Deralakatte, Mangalore 18, India
Amitha M. Hegde
Department of Pedodontics and Preventive Children Dentistry, A.B. Shetty Memorial Institute of Dental Sciences, Deralakatte, Mangalore 18, India
Saurabh Joshi
Department of Pedodontics, Rural Dental College, Loni, Maharashtra, India
European Journal of Dentistry and Medicine
Year: 2012 | Volume: 4 | Issue: 1 | Page No.: 1-7







ABSTRACT
Oral-facial-digital syndrome type 1 (OFD1) is characterized by an X-linked dominant mode of inheritance with lethality in males. It presents with peculiar malformations in the oral cavity and defects in the face and digits of the upper and lower extremities. Accurate diagnosis following clinical investigations by allied health professionals is indispensable to plan out a systematic management protocol in these victims in order to minimize future odontogenic problems. This report presented a unique case of females in a family suffering from manifestations of OFD1 syndrome since three generations. The characteristic clinical features of all the female members were promptly identified, investigated and the inter-disciplinary management protocol that was systematically instituted is depicted in this report.
PDF Abstract XML References Citation
Received: October 11, 2011;
Accepted: October 25, 2011;
Published: March 20, 2012
How to cite this article
Arun M. Xavier, Kavita Rai, Amitha M. Hegde and Saurabh Joshi, 2012. Hereditary Oro-facial Digital Syndrome Type 1: Diagnosis and Management-case Report. European Journal of Dentistry and Medicine, 4: 1-7.
DOI: 10.3923/ejdm.2012.1.7
URL: https://scialert.net/abstract/?doi=ejdm.2012.1.7
DOI: 10.3923/ejdm.2012.1.7
URL: https://scialert.net/abstract/?doi=ejdm.2012.1.7
INTRODUCTION
Oro-facial digital (OFD) syndrome is a generic name for a variety of different genetic disorders that result in malformations of the oro-facial region and digits (King and Sanares, 2002). Based on their distinguishing physical signs, symptoms and mode of inheritance, several OFD syndromes are enlisted in literature (Gorlin and Psaume, 1962; Gorlin et al., 1961; King and Sanares, 2002). OFD1 is a pleiotropic disease crucially associated with dysfunction of the primary cilia (Macca and Franco, 2009). Males with this condition are mostly malformed fetuses delivered by women with OFD1 (Macca and Franco, 2009).
The typical oral and peri-oral features of ODF1 include ankyloglossia, multi-lobulated tongue with the presence of nodules, clefts of the alveolar ridge, multiple hypertrophied frenula, cleft lip-palate, etc. (Mihci et al., 2007). The facial abnormalities most commonly include aplasia of the alar cartilage, ocular hypertelorism, strabismus and alopecia (King and Sanares, 2002; Mihci et al., 2007). Brachydactyly, syndactyly, clinodactyly of the fifth finger commonly, radial and ulnar deviation and duplicated hallux (great toe) are some digital variations (Mihci et al., 2007). Systemically debilitating conditions like polycystic renal disease, sapasmodic movements/tics, brain malformation, delayed motor and speech development may also be present in some patients (Gorlin and Psaume, 1962; King and Sanares, 2002; Mihci et al., 2007).
There is very limited documented dental literature regarding the oral manifestations of OFD1 syndrome and its clinical management in the Asian population. This report presents unique clinical cases of OFD syndrome with hereditary involvement among the female members of a family and the management protocol that was undertaken.
CASE REPORT
Two south Indian female siblings aged 9 and 12 years reported to the Department of Pediatric Dentistry, A.B. Shetty Memorial Institute of Dental sciences, Mangalore with a chief complaint of decay and malaligned teeth (Fig. 1a, b, 2a, b). The children were accompanied by their grandmother, mother and their youngest girl sibling aged 5 years on the first day of visit.
The eldest sibling also complained of an enlarged tongue with noticeable swellings claimed to have increased in size since birth (Fig. 1c). A burning sensation on having hot and spicy food and pain upon palpating the swollen areas was also reported. Accompanying it, frequent bruising of the lateral borders of the tongue upon mastication, where the swellings were appreciably bigger in size was noticed. A detailed medical and family history was recorded and the patients were examined both clinically and radio graphically.
The 12 year old girl had a short stature (143 cm) with shorter upper and lower limbs. The palm (Fig. 1d) and feet of either side (Fig. 1e) were typically small with shorter and wider digits, suggestive of brachydactyly. The first toes in both the feet were larger than the others and resembled a duplicated hallux. The patient was dolichocephalic, leptoprosopic with a convex facial profile.
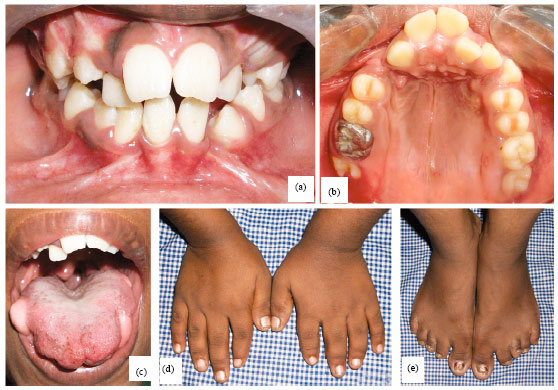 | |
| Fig. 1(a-e): | Clinical features of the 12-year old girl; (a) Anterior view of malaligned dentition, (b) High arched palate, (c) Solitary tongue nodules in the lateral borders, (d) Palms showing small digits and (e) Feet showing small digits |
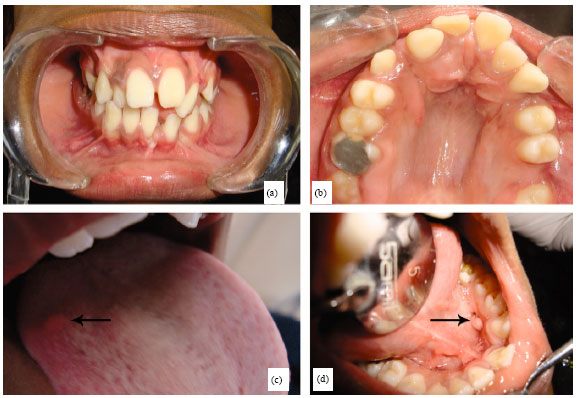 | |
| Fig. 2(a-d): | Clinical features of the 9-year old girl; (a) Anterior view of malaligned dentition, (b) High arched palate, (c) A nodular swelling on the right dorsum of tongue and (d) A suggestive fibromatous mass on the left lower lingual mucosa |
Other extra oral facial features included a broadened forehead, base of nose and widely set eyes. Alopecia was noted at the central portion of the head circumference. Intraoral soft tissue examination revealed macroglossia with superficial and inferior clefting of the anterior 2/3rd of the tongue. Solitary tongue nodules of sizes 1.5x1 cm and lesser were noticed on the lateral borders of the tongue bilaterally (Fig. 1c). The surface of the nodules was smooth, featuring benign mass. Aplasia of papillae over the tongue was also a significant finding. Hard tissue examination revealed clefting of the hypoplastic mandibular alveolar process between 22 and 24 and 31 and 32 (Fig. 1a, b), micrognathia, narrow constricted upper arch, high arched palate (Fig. 1b), as the other striking findings apart from decayed teeth and crowded dentition. Orthopantamograph showed the presence of supernumerary teeth between 12 and 13 and missing teeth in relation to 23 and 32.
All the permanent 1st molars were grossly decayed with pulpal involvement in the 9 year old sibling, with a huge pulp polyp in molar necessitating its extraction. The significant extra oral clinical findings in this 2nd sibling included a broadened forehead, flattened nasal bridge, bilateral downward slanting of the lateral canthi of eyes, a convex facial profile with incompetent lips. Intraorally, accessory frenulum, cleft involving the alveolar process in both the arches, a soft elevated non tender nodular swelling of 2x2 mM size in the right border of the middle 3rd of the tongue (Fig. 2c), narrow and constricted maxillary arch, high arched palate (Fig. 2b) and anterior open bite were the striking features. A 0.5 cm movable, non tender soft tissue fibrous mass, with well defined borders was noticed at the left lower part of the floor of the mouth (Fig. 2d). Orthopantamograph revealed the presence of a supernumerary tooth between 13 and 14, congenitally missing 32 and an impacted left upper lateral incisor.
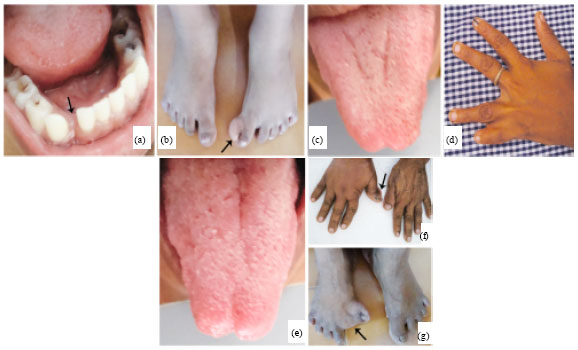 | |
| Fig. 3(a-g): | Clinical features of the other family members 5-year old sibling; (a) Clefting seen in the region of 42 and 43, (b) Duplicated hallux (great toe) in the left leg, Mother, (c) Bifid tongue, (d) Syndactyly of digits in the left hand, Grandmother, (e) Bifid tongue, (f) Abnormal bony swelling in the right thumb and (g) Syndactyly of the great toe in the right leg |
The extra-ordinary clinical presentation of similar extra and intra oral signs in the siblings was suggestive of the possible occurrence of a genetic disorder/syndrome. This urged the need for a thorough clinical examination of the other members who had accompanied them from the family. To our surprise, all the 3 generations had features that had similarities to great extent. Grossly decayed teeth and enamel dysplasia were noticed in the youngest sibling and the members of the other generations. The youngest sibling had clefting of the mandibular alveolar process between 82 and 83 (Fig. 3a), a fibrous movable soft tissue mass of 0.3x0.2 cm in the lingual gingival mucosa and widely set eyes. A duplicated hallux (great toe) was also noticed in the left leg of the 3rd sibling (Fig. 3b). Additionally, syndactyly of digits of either upper or lower limbs (Fig. 3d-f), high arched palate and bifid tongue (Fig. 3c-e) were common to all the 3 of them.
The children were born to non-consanguineous parents and the mother reported of uneventful pregnancies. The mother of these siblings admitted of no history of abortions, male or still birth. The family history revealed that only the maternal grandmother and mother suffered from similar clinical and dental conditions and none of the grandmother’s brothers or her sons had any visible/known defect. The clinical and radiographic features and the family history were consistent with a diagnosis of an X-linked dominant trait of OFD1 with female predisposition and a pedigree chart of the family’s condition was formulated (Fig. 4).
The comprehensive treatment for OFD1 syndrome includes a wide array of measures, as the clinical presentation of the same is diverse. As a part of the treatment plan, the treatment alternatives were explained to the children and their mother. Dietary counseling and education of maintenance of good oral hygiene were delivered.
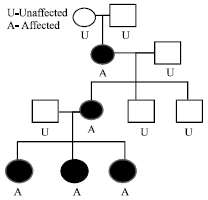 | |
| Fig. 4: | Pedigree representation of OFD1 syndrome in the family |
Root canal treatment was performed on all the permanent 1st molars that were decayed and pulpally involved followed by placement of stainless steel crowns. Light cure glass ionomer and composite restorations for decayed teeth following excavation and cavity preparation was done. Extraction of root stumps, pit and fissure sealants for teeth with incipient carious lesions etc. were also performed in the siblings, according to their needs. The Department of Oral and maxillofacial surgery undertook the surgical debulking of lipomatous swellings on the tongue in the eldest sibling under general anesthesia. The malocclusion of the 9 and 12 year old sisters are being currently managed at the Dept of Orthodontics and Dentofacial Orthopedics.
Surgical orthopedic correction of syndactyly/polydactyly in the other affected members of the family was suggested. As a part of the treatment protocol, genetic counseling to the family members was also provided.
DISCUSSION
The diagnosis of OFD1 is usually established at infancy based on characteristic oral, facial and digital anomalies. A 50% risk of inheriting the disease causing gene has been reported in children of females with OFD1. Most male fetuses of an affected mother may fail to survive or lead to miscarriages (Ferrante et al., 2001). This was however, not noticed in the family studied in this report. The OFD1 gene is assigned to locations Xp 22.3-22.2, or, on the 22nd band of the p arm of the X-chromosome (Ferrante et al., 2001). The familial pattern of inheritance of this X linked dominant trait in the females of the examined family, suggested a definite diagnosis of Oro-facial-digital syndrome Type I. The diagnosis could have been confirmed through molecular genetic testing of samples from all the family members, but wasn’t performed due to the patient’s unwillingness to participate.
A bifid or trifid tongue associated with nodules, usually hamartomatous or lipomatous, is found to occur in 1/3rd the population with OFD1 (Sousa and Kanaan, 1994; King and Sanares, 2002; Mihci et al., 2007). Other common findings include ankyloglossia with short lingual frenulae (King and Sanares, 2002; Mihci et al., 2007). All the members of the current report presented with bifid tongue and the 2 elder siblings presenting with characteristic nodular swellings on the tongue surface.
Abnormalities in the palate including submucous cleft palate, trifurcation of the soft palate, or high arched palate occur in more than half of this victimized group (Al-Qattan, 1998; King and Sanares, 2002). Hyperplastic accessory gingival frenula, extending from the buccal mucous membrane to the alveolar ridge have found to result in alveolar clefts or notching of the alveolar ridge in OFD1 (King and Sanares, 2002; Mihci et al., 2007). Clefting of the alveolar process of the jaw was only noticed in the 3 siblings of our report, while high arched palate and bifid tongue was seen among all the female members studied.
The incidence of dental caries in the 3 generations was significant, the causes being multifactorial (Motlagh et al., 2007; Zeraati and Motlagh, 2006; Razafindrabe et al., 2007; Ogundele and Ogunsile, 2008; Malekipour et al., 2008). Understanding the exact etiology of dental decay and instituting an individualized preventive or corrective strategy can lower the caries prevalence in this high risk group (Mahvi et al., 2006; Madukwe, 2007).
Digital abnormalities like duplicated hallux, brachydactyly and syndactyly of varying degrees of severity was noticed in the present family, as was reported in an earlier published report on OFD1(King and Sanares, 2002). Dry and brittle hair, usually accompanied by partial alopecia in OFD1 syndrome (Del et al., 1999) was noticed in the eldest sibling of the 3rd generation in this report. The short stature in the eldest sibling maybe attributed to familial factors or a severe genetic expression of this condition (Mohammadian and Khoddam, 2007).
In fewer than 50% of individuals with OFD1 syndrome, the occurrence of polycystic kidney disease in later childhood or adulthood has served as a diagnostic marker (Coll et al., 1997). The age of onset is most often in adulthood, but renal cysts in children have been described. Metabolic disturbances associated with kidney disease patients can also lead to chronological enamel hypoplasia of the primary and permanent teeth (Kaya et al., 2008), reflecting to the dysplastic changes in enamel noticed in the female population of this report. However, the siblings in this report were referred to a nephrologist for investigations, who confirmed no existing renal disturbances.
CONCLUSION
The current report observes a severe expression of clinical features related to OFD1 in the 3rd generation of the family than to the earlier generations having fewer findings. Genetic counseling can provide these individuals and families with information on the nature, inheritance and implications of genetic disorders to help them make informed medical and personal decisions. The authors were successful in providing genetic counseling and helped imparting knowledge and insight regarding the family’s medical condition.
REFERENCES
- Al-Qattan, M.M., 1998. Cone-shaped epiphyses in the toes and trifurcation of the soft palate in oral-facial-digital syndrome type-I. Br. J. Plastic Surg., 51: 476-479.
CrossRefDirect Link - Coll, E., R. Torra, J. Pascual, A. Botey and J. Ara et al., 1997. Sporadic orofaciodigital syndrome type I presenting as end-stage renal disease. Nephrol. Dial. Transplant, 12: 1040-1042.
PubMed - Ferrante, M.I., G. Giorgio, S.A. Feather, A. Bulfone and V. Wright et al., 2001. Identification of the gene for oral-facial-digital type I syndrome. Am. J. Hum. Genet., 68: 569-576.
PubMed - Gorlin, R.J., V.E Anderson and C.R. Scott, 1961. Hypertrophied frenuli oligophrenia, familial trembling and anolmalies of the hand. New. Engl. J. Med., 264: 486-489.
PubMed - Gorlin, R.J. and J. Psaume, 1962. Orodigitofacial dysostosis-A new syndrome: A study of 22 cases. J. Pediatr., 61: 520-530.
CrossRefDirect Link - King, N.M. and A.M. Sanares, 2002. Oral-facial-digital syndrome-type I: A case report. J. Clin. Pediatr. Dent., 26: 211-215.
PubMed - Macca, M. and B. Franco, 2009. The molecular basis of oral-facial-digital syndrome type 1. Am. J. Med. Genet. C Semin. Med. Genet., 151: 318-325.
PubMed - Sousa, Y.T.S. and D.D. Kanaan, 1994. The oro-facial-digital syndrome-manifestations in the oral cavity: Case report. Braz. Dent. J., 57: 71-74.
PubMed - Razafindrabe, A.B., V.H. Randriamanantenasoa, M.F. Andrianasolo, J.B. Radaviarison, V.O. Rasoarimasy and J.D. Rakotovao, 2007. Epidemiological and clinical aspects of dental cellulitis in antananarivo. J. Medical Sci., 7: 1108-1111.
CrossRefDirect Link - Kaya, S., N. Hamamci, I. Yavuz, O. Adiguzel and E.C. Tumen, 2008. Oral health and evaluation of skeletal development in children with renal disease. Trends Med. Res., 3: 24-30.
CrossRefDirect Link - Ogundele, B.O. and S.E. Ogunsile, 2008. Dental health knowledge, attitude and practice on the occurrence of dental caries among adolescents in a Local Government Area (LGA) of Oyo State, Nigeria. Asian J. Epidemiol., 1: 64-71.
CrossRefDirect Link - Mohammadian, S. and H. Khoddam, 2007. An etiologic evaluation of children with short stature in Gorgan (Northeast Iran), 2005. J. Medical Sci., 7: 1206-1209.
CrossRefDirect Link - Madukwe, I.U., 2007. Patient`s normative oral health needs in a tertiary dental hospital. J. Medical Sci., 7: 701-703.
CrossRefDirect Link - Motlagh, M.G., G.R.J. Khaniki and H. Adiban, 2007. Investigation of dental caries prevalence among 6-12 year old elementary school children in Andimeshk, Iran. J. Med. Sci., 7: 116-120.
CrossRefDirect Link - Mahvi, A.H., M.A. Zazoli, M. Younecian, B. Nicpour and A. Babapour, 2006. Survey of fluoride concentration in drinking water sources and prevalence of DMFT in the 12 years old students in Behshar City. J. Med. Sci., 6: 658-661.
CrossRefDirect Link - Zeraati, H. and M.G. Motlagh, 2006. An investigation on DMFT and DMFS of first permanent molars in 12 year old blind children in residential institutes for blinds in Tehran, Iran. J. Medi. Sci., 6: 1-4.
CrossRefDirect Link - Malekipour, M.R., M. Messripour and F. Shirani, 2008. Buffering capacity of saliva in patients with active dental caries. Asian J. Biochem., 5: 280-283.
CrossRefDirect Link