
Souravh Bais
Department of Pharmacology, Rayat Institute of Pharmacy, Railmajra, Shaheed Bhagat Singh Nagar District, 144506 Punjab, India
LiveDNA: 91.5733
Renu Kumari
Department of Pharmacology, Rayat Institute of Pharmacy, Railmajra, Shaheed Bhagat Singh Nagar District, 144506 Punjab, India
Yash Prashar
Department of Pharmacology, Rayat Institute of Pharmacy, Railmajra, Shaheed Bhagat Singh Nagar District, 144506 Punjab, India
LiveDNA: 91.15949
Current Research in Neuroscience
Year: 2016 | Volume: 6 | Issue: 1 | Page No.: 1-15







ABSTRACT
Alzheimer’s Disease (AD) is the most common cause of dementia worldwide, characterized as a progressive and irreversible neurodegenerative disease. It is probably caused by complex interactions among multiple genetic, epigenetic and environmental factors. The pathophysiology of AD is largely represented by the neurotoxic events triggered by the proteins like β-amyloid cascade and the hyper phosphorylation of microtubule associated tau proteins and other copathogens in neurons. These processes lead respectively to the formation of neurotic plaques and neurofibrillary tangles which are the pathological hallmarks of the disease. Last 20 years extensive study was done to search (through the internet, books, journals and software’s) available data to elaborate the current update on Alzheimer’s disease. The available data related to etiology, pathophysiology, molecular pathway, traditional cure and available treatment of AD were discussed throughout this study. This study will provide an enough data to various academicians regarding current pharmacological, non-pharmacological interventions, ethnopharmacological treatments and other relevant data related to AD. Due to in availability of proper cure for this disorder and much of the treatment available has been able only delay the progression of the disease or provide symptomatic relief for a short period of time. So much more targeted approach should be discovered to resolve the complications. Arise due to tau proteins and further study on amyloids in underway.
PDF Abstract XML References Citation
Received: June 21, 2016;
Accepted: August 12, 2016;
Published: September 15, 2016
How to cite this article
Souravh Bais, Renu Kumari and Yash Prashar, 2016. A Review on Current Strategies and Future Perspective in Respect to Alzheimer’s Disease Treatment. Current Research in Neuroscience, 6: 1-15.
DOI: 10.3923/crn.2016.1.15
URL: https://scialert.net/abstract/?doi=crn.2016.1.15
DOI: 10.3923/crn.2016.1.15
URL: https://scialert.net/abstract/?doi=crn.2016.1.15
INTRODUCTION
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder scientifically categorised as change in cognition and impairment of memory leads to difficulties in day to day working1. Alzheimer’s disease is the most common form of dementia in the last 60 years. The percentage of persons with Alzheimer disease has been recorded by a factor of two fold increase every 5 years of age, so, it may be frequent as the person gets old. By 2050, the number of cases in US2 is predicted to rise to13.2 million. An IndoUS studies assessed prevalence of Alzheimer disease in a settingof rural India. Few health care systems will be able to cope with this development. These review highlights of the most informative developments in AD study and raises major unresolved issues.
The cognition decline in AD is directly proportional to loss of neurons in specific brain regions3. Although, AD clearly causes loss of neurons in specific brain regions (e.g., of pyramidal cells in lamina II of the entorthinal cortex and in the CA1 region of the campus) much of the overall loss of brain volume appears to be due to the shrinkage, loss of brain volume appears to be due to the shrinkage and loss of neuronal processes. Progressive decreases in cortical thickness can be detected in multiple brain regions by brain Magnetic Resonance Imaging (MRI) in AD patients, correlate with the cognitive decline and predict conversion from Mild Cognitive Impairment (MCI) to AD4,5. The introduction of functional MRI (fMRI) in AD has revealed the alteration in neuronal activities or decreases the risk of disease prognosis in patients. The change in connectivity will remain in default mode due to hyperactivation of the hippocampus during execution of memory tasks5,6.
Another reason is to overstimulation of specific neuronal populations which is so called excitotoxicity and neurodegeneration in AD and related conditions. It is interesting in this regard that AD is associated with an increased incidence of epileptic seizures which is most evident in patients with early-onset forms of the disease7.
Loss of synapses and dendritic spines correlates better with the cognitive decline in AD than loss of neurons8. The different neuronal cultures showed the synaptodendritic refraction and found same with transgenic mice when exposed to AD causing factors9 (Fig. 1).
Taken together, these studies suggest that aberrant neural network activity, dysfunction and loss of synapses and degeneration of specific neuronal population are the main substrates of cognition decline in AD.
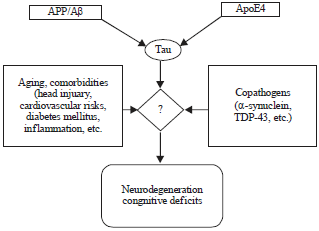 | |
| Fig. 1: | Multifactorial basis of Alzheimer’s disease pathogenesis |
As outlined below, it is likely that these abnormalities are caused by copathogenic interactions among diverse factors and pathways (Fig. 1).
Multifactorial etiology of AD: The AD is very likely caused by complex interactions among multiple genetic, epigenetic and environmental factors.
Genetic factor’s mutations in three genes-amyloid precursor protein (APP): Presenilin PS1 and PS2 cause early-onset (<60 years) Autosomal dominant AD10 which probably accounts for less than 1% of AD cases11. Then mutations affect processing of APP which leads to altered production of different Aβ peptides and their ratio10. Down’s syndrome patients carrying an extra copy of chromosome 21 on which the APP gene resides, develop early-onset dementia with pathological hallmarks of AD in their brains12, consistent with the idea that over expression of APP cause early-onset AD. Moreover, increased APP gene expression caused by genetic variations on the promoter sequence may be a risk factor for late-onset AD with levels of APP expression correlating inversely with age of disease onset13.
Apolipoprotein (Apo) E4: It (used here to refer to either the ApoE ε4 allele or the protein it encodes) has been genetically linked to late-onset (>60 years) familial and sporadic AD which accounts for most AD cases and has a gene-dose effect on increasing the risk and lowering the age of onset of the disease14,15. Recent genome-wide association studies (GWASs) identified that ApoE4 is a major gene associated with the age-related cognitive decline in humans16 which is in line with a longitudinal study showing that age-related memory decline in non-demented ApoE4n carriers diverges from that of non-demented non carriers before the age of 60 years17. Thus, ApoE4’s detrimental effect on cognition occurs before the typical signs of AD arise. In contrast, ApoE2 may protect against AD in some populations15.
Epigenetic factor: Epigenetic mechanism may also play a role in AD pathogenesis18. Studies on human post-mortem brain samples and peripheral leukocytes as well as transgenic animal models have shown that aging and AD are associated with epigenetic dysregulation at various levels including abnormal DNA methylation and histone modifications19. Pharmacological inhibition of DNA methylation in the hippocampus after learning task impaired memory consolidation in mice18 and promotion of histone acetylation improved learning and memory in a mouse model of AD and increased learning-related gene expression in aged wild-type mice20,21, suggesting epigenetic regulation of learning and memory in health and memory in health and disease.
Environmental factor: Aging is the most important known non genetic risk factor for late-onset AD. Potential environmental risk factors for late-onset AD include head injury, low educational levels, hyperlipidaemia, hypertension, homocystinemia, diabetes mellitus and obesity22-25.
Pathophysiology and therapeutic targets for treatment of AD
Aβ and Other APP products: The Aβ peptides derived from APP are the main constituent of amyloid plaques26,27. Over expression of APP in humans through duplication of its gene or trisomy of chromosome 21 causes early-onset of AD28. The catalytic subunit of the γ-secretase protein complex that releases Aβ peptide from its precursor is formed by PS1 or PS2. Autosomal dominant mutations in APP, PS1 or PS2 that alter APP processing and the production or self-aggregation and accumulation of Aβ, promoting aggregation and accumulation of Aβ in the brain causes early-onset AD10. Neural expression of mutant human APP (hAPP) either alone or in combination with mutant PS1 in transgenic rodents causes several AD like alterations which is explained below29-32. The immediate early gene arc which directly binds to PS1 to regulate γ-secretase trafficking is required for neuronal activity-dependent Aβ production33. Results obtained in diverse experimental models suggest that insoluble Aβ fibrils found in amyloid plaques and monomeric Aβ are less pathogenic than soluble, nonfibrillar assemblies of Aβ such as Aβ dimers, trimers and larger oligomers.
ApoE4: The major impact of ApoE4 on AD risk is clearly not understood and less attention, it has received in the field as compared to APP, Aβ, tau and inflammation. The ApoE4 was identified as a genetic risk factor for AD, in vitro and in vivo studies have explored its structural properties and functions in neurobiology, its cellular source-dependent physiological and pathophysiological activities within the brain and its Aβ-dependent and independent roles in AD pathogenesis.
ApoE: Polymorphisms and functions in neurobiology: The ApoE is a polymorphic protein with an important and diverse role in neurobiology. It occurs in three common isoforms (ApoE2, ApoE3 and ApoE4) in humans and differs from one another by single-amino acid substitution9,34-36. The ApoE3 stimulates the neurite outgrowth and ApoE4 inhibits it34-36. In addition, ApoE modulates glutamate receptor recycling in neurons with ApoE3 stimulating and ApoE4 inhibiting this process37.
Cellular source-dependent roles of ApoE4 in AD: Pathogenesis cellular derived ApoE has distinct roles in both physiological and pathophysiological pathways34-36. Astrocytes have long been recognized as the primary source of ApoE in the brain and expression of ApoE in astrocytes is increased during aging and in response to estrogen and activation of liver X receptor or NF-κB34-36. In vitro and in vivo studies suggest that astrocyte-derived ApoE has isoform-specific effects on Aβ clearance or deposition38, a neurite outgrowth39 and behavioural performance.
Aβ-dependent roles of ApoE4 in AD: Pathogenesis in vivo, ApoE is associated with amyloid plaques and in vitro lipid-free ApoE3 and ApoE4 can form stable complexes with Aβ Peptides, with ApoE4 forming complexes more rapidly and effectively34,38. Studies in ApoE-deficient mice expressing mutant hAPP demonstrate that ApoE is actually required for the formation of fibrillar amyloid plaques. In hAPP transgenic mice, human ApoE stimulates Aβ clearance. The ApoE2 and ApoE3 clear more Aβ than ApoE440,41 which may be related to ApoE isoform-dependent effects on astroglial degradation of Aβ deposits. A recent study demonstrated that C-terminally truncated ApoE4 which is found in AD brains, inefficiently clears Aβ and acts in concert with Aβ to elicit neuronal and deficit in transgenic mice42.
Aβ-independent roles of ApoE4 in AD: Pathogenesis in addition, neural stem cells in adult mice express ApoE and ApoE4 impairs adult hippocampal neurogenesis which might also contribute to ApoE4-associated learning and memory deficit. Since, there is no Aβ accumulation in any of these ApoE4 mouse models, these data strongly suggest an Aβ-independent role of ApoE4 in causing a neuronal and behavioural deficit in vivo43,44.
ApoE4-induced impairment of GABAergic interneurons: The ApoE4 knock in mice show an age-dependent decrease in hilar GABAergic interneurons which correlates with the extent of ApoE4-induced impairments of adult hippocampal neurogenesis and with learning and memory deficits. Dysfunction of the GABAergic system may also contribute to cognitive impairment in humans. The AD patients have decreased GABA and somatostatin levels in the brain and CSF and these alterations are more severe in ApoE4 carriers45.
Tau and other copathogens AD: It is associated not only with the abnormal accumulation of amyloid plaques but also with that of NFTs. The NFTs form intracellularly and are made up of primarily of aggregated tau bearing abnormal posttranslational modifications, including increased phosphorylation and acetylation46-48. Tau function primarily to stabilize the microtubules and that are an aggregation in AD causes deficits through a loss-of-function mechanism49. When it is abnormally modified and assumes pathogenic confirmations, tau becomes enriched in dendritic spines where it can interfere with neurotransmission50. Interestingly, tau reduction prevents Aβ from causing a neuronal deficit in cell culture and hAPP transgenic mice49. Thus, while Aβ acts upstream of tau, its adverse effects depend in part on tau. Moreover, tau reduction also prevents ApoE4-dependent neuronal deficits in vitro and in vivo45, pinpointing tau as a key mediator or enabler of both Aβ and ApoE4-dependent pathogenesis.
Approaches for treatment of AD
Approved drugs
Acetyl cholinesterase inhibitors (AChEIs): Degeneration of cholinergic neurons and decrease in Ach levels in neocortex, hippocampus and basal forebrain plays a major role in the pathophysiology of AD. Various therapeutic approaches are proposed to elevate cholinergic transmission like increasing the amount of ACh precursors, blocking hydrolysis with AchE inhibitors, stimulating nicotinic and muscarinic receptors or using or using cholinomimetic substances. Animal and human data suggest that AchEI is the most efficacious drugs for increasing Ach levels in brain and ameliorating symptoms of AD51. The AchEIs are approved for the treatment of mild to moderate AD52.
The AchEIs include donepezil, rivastigmine, galantamine and tacrine-all approved by the U.S. Food and Drug Administration (FDA) for treating AD. The AchEIs have a modest beneficial effect on cognition and memory53 (Table 1).
DonepezilIs: A reversible inhibitor of AchE with a long plasma half-life of 70 h. It is not hepatotoxic54. In vitro studies show that donepezil offers neuro protection by reducing glutamate excitotoxicity, diminishing βA toxicity and consequently increasing cell longevity55. Donepezil show atrophy of the hippocampus in humans which suggests a neuroprotective effect56.
Rivastigmine: It is a reversible AChEI with higher affinity for brain Ach than peripheral Ach. It inhibits both butyrylcholinesterase and AchE57. It has a plasma half-life of 2 h. Rivastigmine is started at a dose of 1.5 mg BD, then increase to a maximum dose of 6 mg BD. Rivastigmine has demonstrated significant treatment effect on the cognitive (thinking and memory), functional (activities of daily living) and behavioural problems that are commonly associated with AD58.
| Table 1: | Clinical pharmacology of cholinesterase inhibitors useful for reducing the signs of Dementia51-55 |
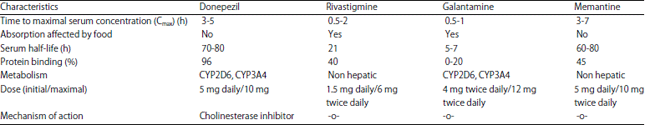 | |
Galantamine: It is a reversible and selective AChEI having 50 times more selectivity for human AchE than for human butyrylcholinesterase. Galantamine also acts as a nicotinic receptor agonist in the brain59. In an animal model, galantamine also increased dopaminergic neurotransmission in the hippocampus60, a brain area and particularly important in memory. A Meta-analysis of 10 randomized, placebo-controlled, double-blind studies concluded that galantamine either improved or prevented the decline of cognition and activities of daily living61.
NMDA receptor antagonist: Glutamate is an excitatory neurotransmitter and acts on a variety of receptors. The NMDA is one such receptor. The NMDA receptor on activation causes potentiating of neuronal activity but in AD, excessive glutamatergic excitotoxicity causes apoptotic cell death and defects in cognition and memory62.
Memantine: An NMDA receptor antagonist has been recently approved by FDA for the treatment of moderate to severe AD, it is found to interfere with the glutamate excitotoxicity63. A study reviews the molecular mechanism of memantine action and the basis for memantine used in AD. Excitotoxic cell death is mediated by over activation on NMDA glutamate receptors which results in excessive ca2+ influx through the receptor associated in the channel. Memantine acts as an uncompetitive and low affinity open channel blocker.
Experimental drugs
Immunization
Anti-amyloid therapy: Anti-amyloid strategies comprise pharmaceutical compounds with distinct mechanisms of action, namely drugs that (i) Facilitate the clearance, (ii) Inhibit the production and (iii) Prevent the aggregation of Aβ64. Many pharmacological compounds have been developed to tackle the "amyloid cascade" with the prospect of reducing the Aβ burden in the brain of mild to moderately demented AD patients65 (Fig. 2, Table 2).
Classification of anti-amyloid therapy: Both active and passive immunization targets the reduction of intracerebral of Aβ load by eliciting humoral response against the Aβ peptide, facilitating its clearance from the brain by immune-mediated mechanisms76. Highly encouraging findings were presented by preclinical studies with transgenic mice with high Aβ load, submitted to active and passive immunization; these strategies proved effective reducing the amount of Aβ in the mouse brain which was supposedly associated with improvements on behaviour and cognition (Fig. 3).
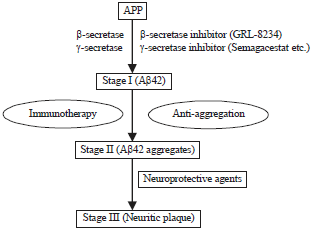 | |
| Fig. 2: | Stages of (Aβ) β-amyloid production with possible targets for treatment44 |
| Table 2: | List of compounds used to target the β-amyloid and act as anti-beta-amyloid treatment in AD |
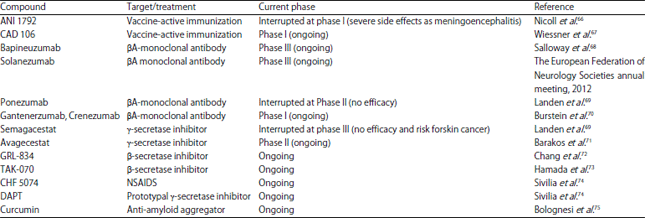 | |
| DAPT: [N-(3, 5-Diflourophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester | |
First generation of immunotherapeutic agents for AD: These agents were based on the active immunization of AD patients with the actual Aβ peptide77. Therefore, this strategy induces an IgM response to generate antibodies against pathogenic Aβ which further mobilize microglia to clean plaques through phagocytosis64,76. Also, the immune response prevents Aβ deposition by removing the excess of soluble Aβ forms from the circulation76.
Second generation of active anti-Aβ Immunotherapeutic agent: It was designed to minimize the risk of eliciting such secondary inflammatory responses or vasogenic oedema by stimulating soluble Aβ derivate immunogens. These vaccines elicit the immune response to raise antibodies against Aβ monomers and oligomers. Studies with the vaccine CAD 106 at phase 1 indicated that it was able to reduce Aβ accumulation in cortical and subcortical brain regions by binding to Aβ aggregates and blocking cellular toxicity with no evidence of micro haemorrhage, vasogenic oedema or inflammatory reactions subsequent to activation of T-cells67. Conversely, passive immunotherapy is based on the intravenous administration of full monoclonal antibodies or antibody fragments from specific exogenous origins which directly target Aβ64.
Several passive immunotherapeutic agents have been evaluated by RCTs over the past years, namely bapinuzumab, solanezumab, gentenerumab, ponezumab and crenezumab. These monoclonal antibodies have high affinity to antigenic determinant epitopes of Aβ, binding either to soluble forms or in plaques, being further recognized by B and T-cells to promote its clearance from the brain.
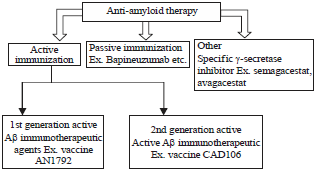 | |
| Fig. 3: | Stages of anti-amyloid therapy64,76 |
In addition, monoclonal antibodies may delay Aβ burden or stop its accumulation within the brain70,77.
Other anti-amyloid strategies have been addressed by clinical trials: Preliminary studies support that the production and accumulation of Aβ. It can be down regulated by the specific γ-secretase inhibitor's like avagacestat and semagacestat69,71.
Semagacestat: A non-selective γ-secretase inhibitor has been examined as potential treatment for AD patients78. Unfortunately, preliminary results showed no efficacy.
Avagacestat: It has been considered as a potentially inhibitor of Ab40 and Ab42 formation with selectivity for effects on APP relative to Notch proteins which interfere with cell proliferation, differentiation and apoptosis. In a study enrolling healthy subjects, this compound exerted a potent selective γ-secretase inhibition with decreased CSF Ab levels as well as the inhibition of the human notch proteins79.
Inhibition of β-secretase: It is another potential mechanism of disease modification in AD given the major role of this enzyme in the amyloidogenic cleavage of the Amyloid Precursor Protein (APP). The BACE-1 (β-site amyloid precursor protein cleaving enzyme 1) produces two peptides (Aβ40 and Aβ42) and its inhibition with specific compounds precludes the excess of amyloid and its accumulation into plaques80.
An inhibitor of β-secretase (GRL-8234) was recently investigated in young transgenic mice with decreased soluble β-amyloid in the brain tissue and with rescued behaviour performance72.
Tau-oriented strategies: Its critical role in pathogenesis of AD, drug development may also target the production, processing (phosphorylation) and aggregation of Tau protein81 (Table 3).
Nicotinic receptor agonist: Another approach to enhance cholinergic function is to administer nicotinic receptor type’s α4β2 and α7 are localized in areas of the brain associated with dementia and memory loss.
| Table 3: | Different drugs which act through tau oriented strategies in AD |
 | |
A selective partial α7 nicotinic receptor agonist 4 OH-GTS-21 is shown to have protective action on cholinergic neurons but not protective for the amyloid over expressing transgenic mice84. A study reported that chronic treatment with RJR-2403 and 17β-estradiol had marked antiamnesic effect in middle-aged ovariectomized rats with experimental Alzheimer type dementia85. A novel compound α-7 nicotinic receptor agonist EVP-6124 is currently in phase II (www.clinicaltrials.gov/NCT01073228). More study is needed to unravel the full potential of nicotinic Ach agonists.
PPAR γ agonists: Peroxisome Proliferator Activated Receptors (PPAR) are a family of nuclear receptors and play an important role in lipid peroxidation, cellular proliferation and differentiation86. The PPAR γ agonists inhibit inflammatory gene expression, alter Aβ homeostasis and exhibit neuroprotective effects87. They also induce apoptotic cell death in glioma cells. A study demonstrated the effect of 15-30 mg of pioglitazone daily in patients with mild AD. The pioglitazone group improved agitation and regional cerebral blood flow in the parietal lobe. The study demonstrated that pioglitazone exhibited cognitive and functional improvement88. It may offer a novel strategy to the already existing treatment paradigm. However, several issues like metabolic effects, genomic effects of PPAR γ agonists need to be addressed.
Antihypertensive drugs
Angiotensin-converting enzyme inhibitors (ACEIs): This reduced inflammation and mental decline in AD patients89 by 50%. Mild-to-moderate AD subjects with blood pressure had a fewer cognitive decline when given an ACE inhibitor that crossed blood-brain barrier (perindopril or captopril) than when given an ACE inhibitor that did not (enalpril or imidapril) or a calcium-channel blocker (nifedipine or nilvadipine)90. A recent study confirmed that ACEIs slow the progression of AD91.
Angiotensin receptor blockers: These block the action of angiotensin II by binding at AT1 receptor sites. They have been reported to reduce AD risk and slow its progression92.
Calcium channel blockers: This is another category of antihypertensive drugs. It may be that β-amyloid, mutations in presenilin proteins or other factors open channel that permit calcium to enter and damage cells93. If so, calcium-channel blockers might be expected to benefit AD patients.
Anti-inflammatory drugs (NSAIDs): Alzheimer’s disease is characterized by neurotic plaques and neurofibrillary tangles. Along with them, there is also evidence of inflammation in the form of cytokines and microglial activation94,95. These observations led to a series of clinical trials with NSAIDs to ascertain their role in Alzheimer’s disease. The mechanism by which NSAIDs affect the pathology of AD is by inhibition of cytokines, decreased platelet aggregation and decrease release of factors which prevent free-radical damage94. A recently published study has tested the effect of NSAID use for more than 5 years on AD. It reported that long-term use of NSAIDs was protective against AD. Maximum effect was seen with the use of Ibuprofen96. The drug treatment reduced the expression of the proinflammatory enzyme COX-2 and iNOS and β-secretase97. Two other studies also demonstrate that ibuprofen reduces microglial activation and cytokine production in transgenic mice over expressing APP98.
Hormones: Insulin has many roles in normal cell functioning. Nasal administration of insulin improved several cognitive measures in subjects with early AD or mild cognitive impairment. Nasal administration allows insulin to reach the brain quickly without affecting insulin levels elsewhere in the body. Nasal administration has also improved verbal memory but only for persons with a specific genetic makeup (apolipoprotein E4 [ApoE ε4] allele)99. Insulin resistance can affect the brain as well as other organs making it difficult for the brain cells to acquire energy for cell maintenance and synaptic connections thus, cell death can occur100. Also, hyperinsulinemia has been found to increase inflammation and βA1-42 in healthy adults101.
Estrogen level: Various pharmacoepidemiological studies have reported that AD is more common in postmenopausal women than men102. These occurrences have led to the hypothesis that estrogen loss in postmenopausal women may contribute to the development of AD. Estrogen is known to reduce the risk of developing dementia. Estrogen is known to modulate ApoE gene, increase the metabolism of APP, protects against oxidative stress and causes direct modulation of neurotransmitters103. Observation data link use of hormone therapy to reduction in Alzheimer’s risk but experimental evidence from clinical trials demonstrates that estrogen increases the incidence of dementia. Several studies are of the view that hormone therapy initiated closer to the time of menopause may reduce the incidence of Alzheimer’s dementia.
Melatonin is a naturally-occurring hormone that is produced in decreasing amounts with age. Melatonin is a powerful antioxidant provides mitochondrial support, protects against tau tangles and reduces βA toxicity. Melatonin readily crosses the BBB and enters all cell structures. A small case study showed 6 mg melatonin daily improved mood and memory over 6 days for 10 patients with mild cognitive impairment104.
Vitamins and minerals: Vitamins B low levels of vitamin B12 and folate appear to be associated with an increased rate of cognitive decline105,106. In addition, for a study of 107 normal individuals, those with low-normal vitamin B12 had the greatest 5 years loss of brain volume. The AD patients typically have high levels of homocysteine107. Researchers have examined the possibility that lowering homocysteine would be therapeutic. A combination of vitamin’s B12 and B6 and folate lowering homocysteine both in normal seniors and in those with mild-to-moderate AD108 but had no effect on cognition. Homocystein levels appear to correlate with aging but not with cognition109.
Vitamin A has received attention because it is essential for learning, memory and cognition because vitamin A levels in the brain decline with age and is lower still in individual with AD110. A metabolic product of vitamin A, retinoic acid is known to slow cell death and offer protection from βA111.
Vitamin E is low in AD patients, a study that followed 3,718 individuals over 6 years examined dietary consumption (excluding vitamin E supplement intake which showed no side effect) of all four tocopherols (α, β, γ and δ) as determined by questionnaires.
Multiple nutrients: Since, AD patients often have multiple deficiencies and it makes sense to use multiple supplements. A study of 14 individuals with early AD found that a multiple formulations (400 μg folic acid, 6 μg vitamin B12, 30 IU vitamin E, 400 mg S-adenosylmethionine, 600 mg N-acetylcysteine and 500 mg acetyl-L-carnitine per tablet with a daily dose of two tablets) improved all measures of cognition although, the increase in memory was not statistically significant. The improvement persisted throughout the 12 months study112.
Lithium is a naturally-occurring mineral found in small amounts in many foods. Lithium increases the level of a neuroprotective protein called bcl-2 in the rat hippocampus and frontal cortex and inhibits glycogen synthase kinase 3β (GSK-3), which is implicated in increasing levels of phosphorylated tau and is bought to be factor leading to βA plaques and cell death113.
Nutrients: Phosphatidylserine (PS) is important in neurotransmission, mitochondria function and cell metabolism. It has also been implicated in the enhancement of a nerve growth factor. In vitro study demonstrates PS increases Ach and provides neuroprotection by inhibiting βA and inflammation114. Supplemental PS was originally derived from bovine brains and study using bovine PS typically found cognitive benefits.
Alpha-lipoic acid (ALA), a fatty acid found in all cells and in some foods is manufactured within the body. It is a powerful antioxidant that readily penetrates the BBB, chelates metals, reduces inflammation and increases Ach. The potential mechanism underlying these and other neuroprotective effects are reviewed elsewhere115.
Omega-3 fatty acids have many beneficial effects that make them investigative prospects for AD. A recent study followed 5,395 healthy adults on an average of 9.6 year to assess the relationship between dietary imega-3 intake and risk of developing AD. Dietary intake of omega-3s was the same for the 365 subjects who developed AD as for those who did not116.
Acetyl-carnitine (ALCAR) is derived from the amino acid L-carnitine studies synergistically with ALA to transport acetyl groups and fatty acids into the mitochondria for energy production. The ALCAR is a small molecule that penetrates the BBB and promotes the biosynthesis of Ach while clearing mitochondria of toxic fatty-acid metabolites117. Its effect on APP helps prevent the build-up of amyloid plaque and preserve synaptic function. The ALCAR also increases the nerve growth factor118.
The ALCAR has been found to produce cognitive benefits for AD patients a small double-blind study of seven probable AD patients and five placebo controls found that 3 g ALCAR daily resulted in less cognitive decline over the course to one year119.
Coenzyme Q10 (CoQ10; Ubiquinone)/Idebenone Coenzyme Q10 is essential for mitochondrial energy production. Mitochondrial dysfunction can result in generation of reactive oxygen species and oxidative stress. Many mitochondrial dysfunctions occur in AD brains including disruption of energy production, apoptosis deregulation, altered calcium homeostasis and others (reviewed elsewhere). For these reasons, mitochondria are viewed as promising therapeutic targets120.
Flavonoids and other plant constituents: HuperzineA (HupA) HupA is an extract from the Chinese moss Huperzia serrata that has been used for centuries in Chinese’s folk medicine to treat a wide range of disease. In vitro and animal studies found HupA preserves Ach longer than tacrine, galantamine or donepezil.
| Table 4: | Different plants for Alzheimer’s therapy reported with neuroprotective.131 |
 | |
The HupA reduces βA-induced neuronal degeneration in the hippocampus and cortex, decrease oxidative damage from cytotoxins and apoptosis induced by βA plaques, protects neurons from βA and free radicals and inhibits glutamate toxicity. A recent meta-analysis of four Chinese studies found 300-500 mcg HupA produced a marked improvement in cognition121-124.
Polyphenols: Curcumin is extracted from the plant curcuma longa (turmeric). Reviewers suggest curcumin may be a promising therapy for AD because it has atleast 10 neuroprotective properties including anti-inflammatory, antioxidant, inhibition of βA formation, clearance of existing βA, copper and iron chelation125. Curcumin readily penetrates the BBB but oral administration may produce barely detectable blood levels at doses of 2 g and low levels at 8 g126. The reasons for bioavailability problems appear to be low absorption, rapid metabolism, quick elimination, the inherent instability and hydrophobic nature of curcumin.
Resveratrol a polyphenols found in red wine, peanuts and other plants reduces oxidative stress, decreases inflammation, reduces βA, protects DNA, decrease cell death and modulates various other systems that protect cells127. Several studies have shown that moderate consumption of red wine reduces the risk of developing AD128. Resveratrol is similar to curcumin in that oral bioavailability is low because it is quickly metabolized and excreted. Attempts have been made to increase bioavailability by the use of quercetin, catechin, apigenin, fisetin, myricetin and kaempferol129. List of different plants used for Alzheimer’s therapy is given in Table 4.
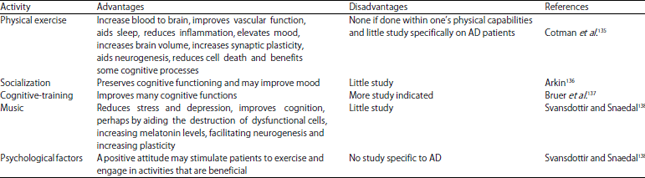
Non-pharmacological or stimulatory therapies for AD: There are several non-pharmacological strategies which manage the functional and behavioural deterioration (www.gmhfonline.org). Physical exercise, cognitive training and socialization are generally thought to facilitate cognitive functioning134 in Table 5.
| Table 5: | Non-pharmacological studies used in AD |
 | |
CONCLUSION
This study was aimed to provide the current and informative data in a compiled frame to treat the AD. It can be concluded that rehabilitation in patients with Alzheimer’s disease should take a multi professional and multidisciplinary study with an emphasis on physiotherapy including enhanced or individually modified physical activity and muscle training.
REFERENCES
- Hebert, L.E., P.A. Scherr, J.L. Bienias, D.A. Bennett and D.A. Evans, 2003. Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Arch. Neurol., 60: 1119-1122.
CrossRefPubMedDirect Link - Gomez-Isla, T., J.L. Price, D.W. McKeel Jr., J.C. Morris, J.H. Growdon and B.T. Hyman, 1996. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J. Neurosci., 16: 4491-4500.
Direct Link - Frisoni, G.B., N.C. Fox, C.R. Jack, P. Scheltens and P.M. Thompson, 2010. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol., 6: 67-77.
CrossRefDirect Link - Putcha, D., M. Brickhouse, K. O'Keefe, C. Sullivan and D. Rentz et al., 2011. Hippocampal hyperactivation associated with cortical thinning in Alzheimer's disease signature regions in non-demented elderly adults. J. Neurosci., 31: 17680-17688.
CrossRefDirect Link - Sperling, R.A., B.C. Dickerson, M. Pihlajamaki, P. Vannini and P.S. LaViolette et al., 2010. Functional alterations in memory networks in early Alzheimer's disease. NeuroMol. Med., 12: 27-43.
CrossRefDirect Link - Palop, J.J. and L. Mucke, 2009. Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol., 66: 435-440.
CrossRefDirect Link - Palop, J.J., J. Chin and L. Mucke, 2006. A network dysfunction perspective on neurodegenerative diseases. Nature, 443: 768-773.
CrossRefDirect Link - Kim, S., J.R. Jensen, K. Cisek, K.E. Funk, S. Naphade, K. Schafer and J. Kuret, 2010. Imaging as a strategy for premortem diagnosis and staging of tauopathies. Curr. Alzheimer Res., 7: 230-234.
CrossRefDirect Link - Bertram, L., C.M. Lill and R.E. Tanzi, 2010. The genetics of Alzheimer disease: Back to the future. Neuron, 68: 270-281.
CrossRefDirect Link - Campion, D., C. Dumanchin, D. Hannequin, B. Dubois and S. Belliard et al., 1999. Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity and mutation spectrum. Am. J. Hum. Genet., 65: 664-670.
CrossRefDirect Link - Sanchez, M.M., S.N. Heyn, D. Das, S. Moghadam, K.J. Martin and A. Salehi, 2012. Neurobiological elements of cognitive dysfunction in down syndrome: Exploring the role of APP. Biol. Psychiatry, 71: 403-409.
CrossRefDirect Link - Brouwers, N., K. Sleegers, S. Engelborghs, V. Bogaerts and S. Serneels et al., 2006. Genetic risk and transcriptional variability of amyloid precursor protein in Alzheimer's disease. Brain, 129: 2984-2991.
CrossRefDirect Link - Corder, E.H., A.M. Saunders, W.J. Strittmatter, D.E. Schmechel and P.C. Gaskell et al., 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science, 261: 921-923.
CrossRefDirect Link - Farrer, L.A., L.A. Cupples, J.L. Haines, B. Hyman and W.A. Kukull et al., 1997. Effects of age, sex and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. J. Am. Med. Assoc., 278: 1349-1356.
CrossRefDirect Link - De Jager, P.L., J.M. Shulman, L.B. Chibnik, B.T. Keenan and T. Raj et al., 2012. A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiol. Aging, 33: 1017-e1-1017-e5.
CrossRefDirect Link - Caselli, R.J., A.C. Dueck, D. Osborne, M.N. Sabbagh and D.J. Connor et al., 2009. Longitudinal modeling of age-related memory decline and the APOE ε4 effect. N. Engl. J. Med., 361: 255-263.
CrossRefDirect Link - Day, J.J. and J.D. Sweatt, 2011. Epigenetic mechanisms in cognition. Neuron, 70: 813-829.
CrossRefDirect Link - Chouliaras, L., B.P.F. Rutten, G. Kenis, O. Peerbooms and P.J. Visser et al., 2010. Epigenetic regulation in the pathophysiology of Alzheimer's disease. Prog. Neurobiol., 90: 498-510.
CrossRefDirect Link - Fischer, A., F. Sananbenesi, X. Wang, M. Dobbin and L.H. Tsai, 2007. Recovery of learning and memory is associated with chromatin remodelling. Nature, 447: 178-182.
CrossRefDirect Link - Peleg, S., F. Sananbenesi, A. Zovoilis, S. Burkhardt and S. Bahari-Javan et al., 2010. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science, 328: 753-756.
CrossRefDirect Link - Barnes, D.E. and K. Yaffe, 2011. The projected effect of risk factor reduction on Alzheimer's disease prevalence. Lancet Neurol., 10: 819-828.
CrossRefDirect Link - Rosendorff, C., M.S. Beeri and J.M. Silverman, 2007. Cardiovascular risk factors for Alzheimer's disease. Am. J. Gariatr. Cardiol., 16: 143-149.
CrossRefDirect Link - Sharp, E.S. and M. Gatz, 2011. The relationship between education and dementia: An updated systematic review. Alzheimer Dis. Assoc. Disord., 25: 289-304.
CrossRefDirect Link - Van den Heuvel, C., E. Thornton and R. Vink, 2007. Traumatic brain injury and Alzheimer's disease: A review. Progr. Brain Res., 161: 303-316.
CrossRefDirect Link - Citron, M., 2010. Alzheimer's disease: Strategies for disease modification. Nat. Rev. Drug Discov., 9: 387-398.
CrossRefDirect Link - De Strooper, B., R. Vassar and T. Golde, 2010. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol., 6: 99-107.
CrossRefDirect Link - Prasher, V.P., M.J. Farrer, A.M. Kessling, E.M.C. Fisher, R.J. West, P.C. Barber and A.C. Butler, 1998. Molecular mapping of Alzheimer-type dementia in Down's syndrome. Ann. Neurol., 43: 380-383.
CrossRefDirect Link - Ashe, K.H. and K.R. Zahs, 2010. Probing the biology of Alzheimer's disease in mice. Neuron, 66: 631-645.
CrossRefDirect Link - Jucker, M., 2010. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat. Med., 16: 1210-1214.
CrossRefDirect Link - Palop, J.J. and L. Mucke, 2010. Amyloid-β-induced neuronal dysfunction in Alzheimer's disease: From synapses toward neural networks. Nat. Neurosci., 13: 812-818.
CrossRefDirect Link - Querfurth, H.W. and F.M. LaFerla, 2010. Alzheimer's disease. N. Engl. J. Med., 362: 329-344.
CrossRefPubMedDirect Link - Wu, J., R.S. Petralia, H. Kurushima, H. Patel and M.Y. Jung et al., 2011. Arc/Arg3.1 Regulates an endosomal pathway essential for activity-dependent β-amyloid generation. Cell, 147: 615-628.
CrossRefDirect Link - Mahley, R.W., K.H. Weisgraber and Y. Huang, 2006. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc. Natl. Acad. Sci. USA., 103: 5644-5651.
CrossRefDirect Link - Huang, Y., 2006. Molecular and cellular mechanisms of apolipoprotein E4 neurotoxicity and potential therapeutic strategies. Curr. Opin. Drug Discov. Dev., 9: 627-641.
PubMedDirect Link - Huang, Y., 2010. Aβ-independent roles of apolipoprotein E4 in the pathogenesis of Alzheimer's disease. Trends Mol. Med., 16: 287-294.
CrossRefDirect Link - Chen, Y., M.S. Durakoglugil, X. Xian and J. Herz, 2010. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. USA., 107: 12011-12016.
CrossRefDirect Link - Kim, J., J.M. Basak and D.M. Holtzman, 2009. The role of apolipoprotein E in Alzheimer's disease. Neuron, 63: 287-303.
CrossRefDirect Link - Holtzman, D.M., R.E. Pitas, J. Kilbridge, B. Nathan, R.W. Mahley, G. Bu and A.L. Schwartz, 1995. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc. Natl. Acad. Sci., 92: 9480-9484.
Direct Link - Bales, K.R., T. Verina, D.J. Cummins, Y. Du and R.C. Dodel et al., 1999. Apolipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci., 96: 15233-15238.
CrossRefDirect Link - Holtzman, D.M., K.R. Bales, T. Tenkova, A.M. Fagan and M. Parsadanian et al., 2000. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA., 97: 2892-2897.
CrossRefDirect Link - Bien-Ly, N., Y. Andrews-Zwilling, Q. Xu, A. Bernardo, C. Wang and Y. Huang, 2011. C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-β (Aβ) and acts in concert with Aβ to elicit neuronal and behavioral deficits in mice. Proc. Natl. Acad. Sci. USA., 108: 4236-4241.
CrossRefDirect Link - Filippini, N., B.J. MacIntosh, M.G. Hough, G.M. Goodwin and G.B. Frisoni et al., 2009. Distinct patterns of brain activity in young carriers of the APOE-ε4 allele. Proc. Natl. Acad. Sci. USA., 106: 7209-7214.
CrossRefDirect Link - Braak, H. and K. Del Tredici, 2011. Alzheimer's pathogenesis: Is there neuron-to-neuron propagation? Acta Neuropathol., 121: 589-595.
CrossRefDirect Link - Andrews-Zwilling, Y., N. Bien-Ly, Q. Xu, G. Li and A. Bernardo et al., 2010. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neusci., 30: 13707-13717.
CrossRefDirect Link - Cohen, T.J., J.L. Guo, D.E. Hurtado, L.K. Kwong, I.P. Mills, J.Q. Trojanowski and V.M.Y. Lee, 2011. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun., Vol. 2.
CrossRefDirect Link - Iqbal, K., F. Liu, C.X. Gong and I. Grundke-Iqbal, 2010. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res., 7: 656-664.
CrossRefDirect Link - Min, S.W., S.H. Cho, Y. Zhou, S. Schroeder and V. Haroutunian et al., 2010. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron, 67: 953-966.
CrossRefDirect Link - Morris, M., S. Maeda, K. Vossel and L. Mucke, 2011. The many faces of tau. Neuron, 70: 410-426.
CrossRefDirect Link - Hoover, B.R., M.N. Reed, J. Su, R.D. Penrod and L.A. Kotilinek et al., 2010. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron, 68: 1067-1081.
CrossRefDirect Link - Krall, W.J., J.J. Sramek and N.R. Cutler, 1999. Cholinesterase inhibitors: A therapeutic strategy for Alzheimer disease. Ann. Pharmacother., 33: 441-450.
CrossRefDirect Link - Small, G.W., P.V. Rabins, P.P. Barry, N.S. Buckholtz and S.T. DeKosky et al., 1997. Diagnosis and treatment of alzheimer disease and related disorders: Consensus statement of the american association for geriatric psychiatry, the Alzheimer's association and the American geriatrics society. J. Am. Med. Assoc., 278: 1363-1371.
CrossRefDirect Link - Hansen, R.A., G. Gartlehner, A.P. Webb, L.C. Morgan, C.G. Moore and D.E. Jonas, 2008. Efficacy and safety of donepezil, galantamine and rivastigmine for the treatment of Alzheimer's disease: A systematic review and meta-analysis. Clin. Interv Aging, 3: 211-225.
PubMedDirect Link - Kosasa, T., Y. Kuriya, K. Matsui and Y. Yamanishi, 2000. Inhibitory effect of orally administered donepezil hydrochloride (E2020), a novel treatment for Alzheimer's disease, on cholinesterase activity in rats. Eur. J. Pharmacol., 389: 173-179.
CrossRefDirect Link - Takada, Y., A. Yonezawa, T. Kume, H. Katsuki, S. Kaneko, H. Sugimoto and A. Akaike, 2003. Nicotinic acetylcholine receptor-mediated neuroprotection by donepezil against glutamate neurotoxicity in rat cortical neurons. J. Pharmacol. Exp. Ther., 306: 772-777.
CrossRefDirect Link - Hashimoto, M., H. Kazui, K. Matsumoto, Y. Nakano, M. Yasuda and E. Mori, 2005. Does donepezil treatment slow the progression of hippocampal atrophy in patients with Alzheimer's disease? Am. J. Psychiatry, 162: 676-682.
CrossRefDirect Link - Camps, P. and D. Munoz-Torrero, 2002. Cholinergic drugs in pharmacotherapy of Alzheimers disease. Mini. Rev. Med. Chem., 2: 11-25.
CrossRefDirect Link - Finkel, S.I., 2004. Effects of rivastigmine on behavioral and psychological symptoms of dementia in Alzheimer's disease. Clin. Therapeut., 26: 980-990.
CrossRefDirect Link - Coyle, J. and P. Kershaw, 2001. Galantamine, a cholinesterase inhibitor that allosterically modulates nicotinic receptors: Effects on the course of Alzheimer's disease. Biol. Psychiatry, 49: 289-299.
CrossRefDirect Link - Wang, D., Y. Noda, Y. Zhou, A. Mouri and H. Mizoguchi et al., 2007. The allosteric potentiation of nicotinic acetylcholine receptors by galantamine ameliorates the cognitive dysfunction in beta amyloid25-35 I.c.v.-injected mice: Involvement of dopaminergic systems. Neuropsychopharmacology, 32: 1261-1271.
CrossRefDirect Link - Loy, C. and L. Schneider, 2006. Galantamine for Alzheimer's disease and mild cognitive impairment. Cochrane Database Syst. Rev.
CrossRefDirect Link - Danysz, W. and C.G. Parsons, 2003. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer's disease: Preclinical evidence. Int. J. Geriatr. Psychiatry, 18: S23-S32.
CrossRefPubMedDirect Link - Parsons, C.G., W. Danysz and G. Quack, 1999. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist-a review of preclinical data. Neuropharmacology, 38: 735-767.
CrossRefDirect Link - Salomone, S., F. Caraci, G.M. Leggio, J. Fedotova and F. Drago, 2012. New pharmacological strategies for treatment of Alzheimer's disease: Focus on disease modifying drugs. Br. J. Clin. Pharmacol., 73: 504-517.
CrossRefDirect Link - Hardy, J. and D.J. Selkoe, 2002. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science, 297: 353-356.
CrossRefPubMedDirect Link - Nicoll, J.A.R., D. Wilkinson, C. Holmes, P. Steart, H. Markham and R.O. Weller, 2003. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: A case report. Nat. Med., 9: 448-452.
CrossRefDirect Link - Wiessner, C., K.H. Wiederhold, A.C. Tissot, P. Frey and S. Danner et al., 2011. The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J. Neurosci., 31: 9323-9331.
CrossRefDirect Link - Salloway, S., R. Sperling, S. Gilman, N.C. Fox and K. Blennow et al., 2009. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology, 73: 2061-2070.
CrossRefDirect Link - Landen, J.W., Q. Zhao, S. Cohen, M. Borrie and M. Woodward et al., 2013. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild-to-moderate Alzheimer disease: A phase I, randomized, placebo-controlled, double-blind, dose-escalation study. Clin. Neuropharmacol., 36: 14-23.
CrossRefDirect Link - Burstein, A.H., Q. Zhao, J. Ross, S. Styren and J.W. Landen et al., 2013. Safety and pharmacology of ponezumab (PF-04360365) after a single 10-minute intravenous infusion in subjects with mild to moderate Alzheimer disease. Clin. Neuropharmacol., 36: 8-13.
CrossRefDirect Link - Barakos, J., C. Carlson, W. Estergard, J. Oh, J. Suhy, C. Jack, E. Siemers, 2011. Vasogenic edema in the setting of β-amyloid lowering therapy, adverse event: What is it and how is it detected? Alzheimer's Dementia, 7: e75-e75.
CrossRefDirect Link - Chang, W.P., X. Huang, D. Downs, J.R. Cirrito and G. Koelsch et al., 2011. β-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J., 25: 775-784.
CrossRefDirect Link - Hamada, Y. and Y. Kiso, 2013. Advances in the identification of β-secretase inhibitors. Expert Opin. Drug Discov., 8: 709-731.
CrossRefDirect Link - Sivilia, S., L. Lorenzini, A. Giuliani, M. Gusciglio and M. Fernandez et al., 2013. Multi-target action of the novel anti-Alzheimer compound CHF5074: In vivo study of long term treatment in Tg2576 mice. BMC Neurosci., Vol. 14.
CrossRefDirect Link - Bolognesi, M.L., M. Bartolini, A. Tarozzi, F. Morroni and F. Lizzi et al., 2011. Multitargeted drugs discovery: Balancing anti-amyloid and anticholinesterase capacity in a single chemical entity. Bioorg. Med. Chem. Lett., 21: 2655-2658.
CrossRefDirect Link - Panza, F., V. Frisardi, B.P. Imbimbo, D. Seripa and F. Paris et al., 2011. Anti-β-amyloid immunotherapy for Alzheimers disease: Focus on bapineuzumab. Curr. Alzheimer Res., 8: 808-817.
CrossRefDirect Link - Bard, F., M. Fox, S. Friedrich, P. Seubert, D. Schenk, G.G. Kinney and T. Yednock, 2012. Sustained levels of antibodies against Aβ in amyloid-rich regions of the CNS following intravenous dosing in human APP transgenic mice. Exp. Neurol., 238: 38-43.
CrossRefDirect Link - Albright, C.F., R.C. Dockens, J.E. Meredith, R.E. Olson and R. Slemmon et al., 2013. Pharmacodynamics of selective inhibition of γ-secretase by avagacestat. J. Pharmacol. Exp. Therapeut., 344: 686-695.
CrossRefDirect Link - Ozudogru, S.N. and C.F. Lippa, 2012. Disease modifying drugs targeting β-amyloid. Am. J. Alzheimer's Dis. Dement., 27: 296-300.
CrossRefDirect Link - Hanger, D.P., B.H. Anderton and W. Noble, 2009. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med., 15: 112-119.
CrossRefDirect Link - Takashima, A., T. Honda, K. Yasutake, G. Michel and O. Murayama et al., 1998. Activation of tau protein kinase I/glycogen synthase kinase-3β by amyloid β peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res., 31: 317-323.
CrossRefDirect Link - Tariot, P.N. and P.S. Aisen, 2009. Can lithium or valproate untie tangles in Alzheimer's disease? J. Clin. Psychiat., 70: 919-921.
PubMedDirect Link - Ren, K., M.A. King, J. Liu, J. Siemann and M. Altman et al., 2007. The α7 nicotinic receptor agonist 4OH-GTS-21 protects axotomized septohippocampal cholinergic neurons in wild type but not amyloid-overexpressing transgenic mice. Neuroscience, 148: 230-237.
CrossRefDirect Link - Sapronov, N.S., Y.O. Fedotova and N.N. Kuznetsova, 2006. Antiamnestic effect of α7-nicotinic receptor agonist RJR-2403 in middle-aged ovariectomized rats with Alzheimer type dementia. Bull. Exp. Biol. Med., 142: 700-702.
CrossRefDirect Link - Willson, T.M., P.J. Brown, D.D. Sternbach and B.R. Henke, 2000. The PPARs: From orphan receptors to drug discovery. J. Med. Chem., 43: 527-550.
CrossRefDirect Link - Landreth, G., 2007. Therapeutic use of agonists of the nuclear receptor PPARγ in Alzheimer's disease. Curr. Alzheimer. Res., 4: 159-164.
CrossRefPubMedDirect Link - Sato, T., H. Hanyu, K. Hirao, H. Kanetaka, H. Sakurai and T. Iwamoto, 2011. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging, 32: 1626-1633.
CrossRefPubMedDirect Link - Ohrui, T., N. Tomita, T. Sato-Nakagawa, T. Matsui and M. Maruyama et al., 2004. Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology, 63: 1324-1325.
Direct Link - Hajjar, I.M., M. Keown, P. Lewis and A. Almor, 2008. Angiotensin converting enzyme inhibitors and cognitive and functional decline in patients with Alzheimer's disease: An observational study. Am. J. Alzheimer's Dis. Dement., 23: 77-83.
CrossRefDirect Link - Trenkwalder, P., 2006. The study on cognition and prognosis in the elderly (SCOPE)-recent analyses. J. Hypert., 24: S107-S114.
CrossRefDirect Link - Small, D.H., R. Gasperini, A.J. Vincent, A.C. Hung and L. Foa, 2009. The role of Aβ-induced calcium dysregulation in the pathogenesis of Alzheimer's disease. J. Alzheimer's Dis., 16: 225-233.
CrossRefDirect Link - Flynn, B.L. and K.A. Theesen, 1999. Pharmacologic management of Alzheimer disease part III: Nonsteroidal antiinflammatory drugs-emerging protective evidence? Ann. Pharmacother., 33: 840-849.
CrossRefDirect Link - Vlad, S.C., D.R. Miller, N.W. Kowall and D.T. Felson, 2008. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology, 70: 1672-1677.
Direct Link - Heneka, M.T., M. Sastre, L. Dumitrescu-Ozimek, A. Hanke and I. Dewachter et al., 2005. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1-42 levels in APPV717I transgenic mice. Brain, 128: 1442-1453.
CrossRefDirect Link - Lim, G.P., F. Yang, T. Chu, P. Chen and W. Beech et al., 2000. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J. Neurosci., 20: 5709-5714.
Direct Link - Reger, M.A., G.S. Watson, P.S. Green, C.W. Wilkinson and L.D. Baker et al., 2008. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology, 70: 440-448.
PubMedDirect Link - Neumann, K.F., L. Rojo, L.P. Navarrete, G. Farias, P. Reyes and R.B. Maccioni, 2008. Insulin resistance and Alzheimer's disease: Molecular links and clinical implications. Curr. Alzheimer Res., 5: 438-447.
Direct Link - Fishel, M.A., G.S. Watson, T.J. Montine, Q. Wang and P.S. Green et al., 2005. Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults. Arch. Neurol., 62: 1539-1544.
CrossRefDirect Link - Henderson, V.W., 1997. The epidemiology of estrogen replacement therapy and Alzheimer's disease. Neurology, 48: 27S-35S.
CrossRefDirect Link - Yaffe, K., G. Sawaya, I. Lieberburg and D. Grady, 1998. Estrogen therapy in postmenopausal women: Effects on cognitive function and dementia. J. Am. Med. Assoc., 279: 688-695.
CrossRefDirect Link - Jean-Louis, G., H. Gizycki and F. Zizi, 1998. Melatonin effects on sleep, mood, and cognition in elderly with mild cognitive impairment. J. Pineal Res., 25: 177-183.
CrossRefDirect Link - Tucker, K.L., N. Qiao, T. Scott, I. Rosenberg and A. Spiro III, 2005. High homocysteine and low B vitamins predict cognitive decline in aging men: The Veterans affairs normative aging study. Am. J. Clin. Nutr., 82: 627-635.
PubMedDirect Link - Morris, M.S., P.F. Jacques, I.H. Rosengerg and J. Selhub, 2007. Folate and vitamin B-12 status in relation to anemia, macrocytosis and cognitive impairment in older Americans in the age of folic acid fortification. Am. J. Clin. Nutr., 85: 193-200.
PubMedDirect Link - McIlroy, S.P., K.B. Dynan, J.T. Lawson, C.C. Patterson and A.P. Passmore, 2002. Moderately elevated plasma homocysteine, methylenetetrahydrofolate reductase genotype and risk for stroke, vascular dementia and Alzheimer disease in Northern Ireland. Stroke, 33: 2351-2356.
CrossRefPubMedDirect Link - Aisen, P.S., L.S. Schneider, M. Sano, R. Diaz-Arrastia and C.H. van Dyck et al., 2008. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. J. Am. Med. Assoc., 300: 1774-1783.
CrossRefDirect Link - Li, L., D. Cao, R. Desmond, A. Rahman, J.J. Lah, A.I. Levey and E. Zamrini, 2008. Cognitive performance and plasma levels of homocysteine, vitamin B12, folate and lipids in patients with Alzheimer disease. Dementia Geriatric Cognit. Disord., 26: 384-390.
CrossRefDirect Link - Goodman, A.B., 2006. Retinoid receptors, transporters and metabolizers as therapeutic targets in late onset Alzheimer disease. J. Cell. Physiol., 209: 598-603.
CrossRefDirect Link - Sahin, M., S.B. Karauzum, G. Perry, M.A. Smith and Y. Aliciguzel, 2005. Retinoic acid isomers protect hippocampal neurons from amyloid-β induced neurodegeneration. Neurotoxicity Res., 7: 243-250.
CrossRefPubMedDirect Link - Chan, A., J. Paskavitz, R. Remington, S. Rasmussen and T.B. Shea, 2009. Efficacy of a vitamin/nutriceutical formulation for early-stage Alzheimer's disease: A 1-year, open-label pilot study with an 16-month caregiver extension. Am. J. Alzheimer's Dis. Other Dementias, 23: 571-585.
CrossRefDirect Link - Martinez, A. and D.I. Perez, 2008. GSK-3 inhibitors: A ray of hope for the treatment of Alzheimer's disease? J. Alzheimer's Dis., 15: 181-191.
Direct Link - Hashioka, S., Y.H. Han, S. Fujii, T. Kato and A. Monji et al., 2007. Phosphatidylserine and phosphatidylcholine-containing liposomes inhibit amyloid β and interferon-γ-induced microglial activation. Free Radic. Biol. Med., 42: 945-954.
CrossRefDirect Link - Holmquist, L., G. Stuchbury, K. Berbaum, S. Muscat and S. Young et al., 2007. Lipoic acid as a novel treatment for Alzheimer's disease and related dementias. Pharmacol. Therapeut., 113: 154-164.
CrossRefDirect Link - Devore, E.E., F. Grodstein, F.J. van Rooij, A. Hofman and B. Rosner et al., 2009. Dietary intake of fish and omega-3 fatty acids in relation to long-term dementia risk. Am. J. Clin. Nutr., 90: 170-176.
CrossRefDirect Link - Carta, A., M. Calvani, D. Bravi and S.N. Bhuachalla, 1993. Acetyl‐L‐carnitine and Alzheimer's disease: Pharmacological considerations beyond the cholinergic sphere. Ann. N. Y. Acad. Sci., 695: 324-326.
CrossRefDirect Link - Taglialatela, G., D. Navarra, R. Cruciani, M.T. Ramacci, G.S. Alema and L. Angelucci, 1994. Acetyl-L-carnitine treatment increases nerve growth factor levels and choline acetyltransferase activity in the central nervous system of aged rats. Exp. Gerontol., 29: 55-66.
CrossRefDirect Link - Pettegrew, J.W., W.E. Klunk, K. Panchalingam, J.N. Kanfer and R.J. McClure, 1995. Clinical and neurochemical effects of acetyl-L-carnitine in Alzheimer's disease. Neurobiol. Aging, 16: 1-4.
CrossRefDirect Link - Bonda, D.J., X. Wang, K.A. Gustaw-Rothenberg, G. Perry, M.A. Smith and X. Zhu, 2009. Mitochondrial drugs for Alzheimer disease. Pharmaceuticals, 2: 287-298.
CrossRefDirect Link - Wang, B.S., H. Wang, Z.H. Wei, Y.Y. Song, L. Zhang and H.Z. Chen, 2009. Efficacy and safety of natural acetylcholinesterase inhibitor huperzine A in the treatment of Alzheimer's disease: An updated meta-analysis. J. Neural Transmission, 116: 457-465.
CrossRefDirect Link - Bais, S., N.S. Gill, N. Rana and S. Shandil, 2014. A phytopharmacological review on a medicinal plant: Juniperus communis. Int. Scholarly Res. Notices, Vol. 2014.
CrossRefDirect Link - Bais, S., N.S. Gill and N. Rana, 2014. Effect of Juniperus communis extract on reserpine induced catalepsy. Inventi Impact: Ethnopharmacol., 2014: 117-120.
Direct Link - Walker, D. and L.F. Lue, 2007. Anti-inflammatory and immune therapy for Alzheimer's disease: Current status and future directions. Curr. Neuropharmacol., 5: 232-243.
CrossRefDirect Link - Ringman, J.M., S.A. Frautschy, G.M. Cole, D.L. Masterman and J.L. Cummings, 2005. A potential role of the curry spice curcumin in Alzheimer's disease. Curr. Alzheimer Res., 2: 131-136.
PubMedDirect Link - Mishra, S. and K. Palanivelu, 2008. The effect of curcumin (turmeric) on Alzheimer's disease: An overview. Ann. Indian Acad. Neurol., 11: 13-19.
CrossRefDirect Link - Luchsinger, J.A., M.X. Tang, M. Siddiqui, S. Shea and R. Mayeux, 2004. Alcohol intake and risk of dementia. J. Am. Geriatrics Soc., 52: 540-546.
CrossRefPubMedDirect Link - De Santi, C., A. Pietrabissa, F. Mosca and G.M. Pacifici, 2000. Glucuronidation of resveratrol, a natural product present in grape and wine, in the human liver. Xenobiotica, 30: 1047-1054.
CrossRefDirect Link - Singh, V., 2015. An ancient approach but turning into future potential source of therapeutics in Alzheimer's disease. Int. Res. J. Pharm., 6: 10-21.
Direct Link - Singh, N., B.R. Pandey and P. Verma, 2011. An overview of phytotherapeutic approach in prevention and treatment of Alzheimer's syndrome and dementia. Int. J. Pharmaceut. Sci. Drug Res., 3: 162-172.
Direct Link - Bais, S. and Y. Prashar, 2015. Identification and characterization of amentoflavone from six species of Juniperus against H2O2 induced oxidative damage in human erythrocytes and leucocytes. Res. J. Phytochem., 9: 41-55.
CrossRefDirect Link - Kakkar, S. and S. Bais, 2014. A review on protocatechuic acid and its pharmacological potential. ISRN Pharmacol.
CrossRefDirect Link - Massoud, F., S. Belleville, H. Bergman, J. Kirk and H. Chertkow et al., 2007. Mild cognitive impairment and cognitive impairment, no dementia: Part B, therapy. Alzheimer's Dementia, 3: 283-291.
CrossRefDirect Link - Cotman, C.W., N.C. Berchtold and L.A. Christie, 2007. Exercise builds brain health: Key roles of growth factor cascades and inflammation. Trends Neurosci., 30: 464-472.
CrossRefDirect Link - Arkin, S., 2007. Language-enriched exercise plus socialization slows cognitive decline in Alzheimer's disease. Am. J. Alzheimer's Dis. Other Dementias, 22: 62-77.
CrossRefDirect Link - Bruer, R.A., E. Spitznagel and C.R. Cloninger, 2007. The temporal limits of cognitive change from music therapy in elderly persons with dementia or dementia-like cognitive nmpairment: A randomized controlled trial. J. Music Therapy, 44: 308-328.
CrossRefDirect Link - Svansdottir, H.B. and J. Snaedal, 2006. Music therapy in moderate and severe dementia of Alzheimer's type: A case-control study. Int. Psychogeriatr., 18: 613-621.
CrossRefDirect Link