
Mahin B. Izadi
Not Available
S. Ali Moosawi-Jorf
Not Available
Asian Journal of Plant Sciences
Year: 2007 | Volume: 6 | Issue: 7 | Page No.: 1137-1142







ABSTRACT
Teliospores of Ustilago scitaminea were collected from whips of infected sugarcanes and cultured on YGC medium which conducted to isolation of yeast-like and mycelial colonies. White mycelia were also isolated from culturing of pieces of infected tissues of sugarcane. Results of PCR using specific primers bE4 and bE8 revealed the fungal detection in extracted DNA from yeast-like and mycelial colonies. PCR reaction was specifically amplified a DNA fragment of 495 bp in all three types of colonies of yeast-like and mycelial forms. This DNA fragment was not amplified in DNA extraction of yeast-bacterial colonies and other saprophytic-mycelial fungi.
PDF Abstract XML References Citation
How to cite this article
Mahin B. Izadi and S. Ali Moosawi-Jorf, 2007. Isolation and Identification of Yeast-Like and Mycelial Colonies of Ustilago scitaminea Using Specific Primers. Asian Journal of Plant Sciences, 6: 1137-1142.
DOI: 10.3923/ajps.2007.1137.1142
URL: https://scialert.net/abstract/?doi=ajps.2007.1137.1142
DOI: 10.3923/ajps.2007.1137.1142
URL: https://scialert.net/abstract/?doi=ajps.2007.1137.1142
INTRODUCTION
Sugarcane smut was first reported from Natal in South Africa (McMartin, 1945). Ustilago scitaminea, was reported to be the causal agent of sugarcane smut and it belongs to class Basidiomycetes, subclass Ustilaginomycetidae, order Ustilaginales, family Ustilaginaceae, includes a group of phytopathogenic fungi (Vanky, 1999).
In Iran, the disease was reported in the Haft-Tappeh region in 1971 and was associated with specific problems in sugarcane production (Ershad and Bani-Abbasi, 1971; Moosawi-Jorf et al., 2006; Moosawi-Jorf and Izadi, 2007). The causal agent of sugarcane smut in Iran was firstly described and identified on smutted plants of Erianthus vavenae as Ustilago sacchari rabenh (Ershad and Bani-Abbasi, 1971); but Sydow (Ricaud et al., 1989) declared that the smut on Erianthus vavenae differ with sugarcane smut and suggested the subspecies Ustilago scitaminea as the determinant fungi of sugarcane smut.
At the teliospore germination time of Ustilago scitaminea, spores extend in size, split its exosporidium (external wall of teliospores) and produce small hyphae called promycelium. Each cell of promycelium produces a sporidium (Alexander and Krishna, 1978). Germination of sporidia leads to the production of germ tube or same sporidial cells (Chona, 1943). Surface infection of teliospores with Fusarium moniliforme var. subglatinans reduced its germination considerably (Sinha and Singh, 1983).
Heterothallism in Ustilago scitaminea is bipolar. Sporidia belongs to two mating types; plus (+) and minus (-). Single haploid (n) sporidia continue to multiply by budding in a yeast-like manner. Mycelial dikaryotic (n+n) phase appears following dikaryotization that occur by one of the following ways: (a) fusion of compatible sporidia (b) promycelial anastomosis; The growth of two promycelium together and lysis of cell wall in crossed site; finally the migration of the nucleus to fused cell and (c) hyphal anastomosis of two mycelia of different mating types arising from sporidial germination (Agnitori, 1990; Moosawi-Jorf et al., 2006).
Three kinds of colonies of Ustilago scitaminea were identified; complex colonies cream in color, white mycelial colonies and yeast-like colonies with no clear correlation between them. It had been suggested that they interrelate in position and ploidy of nucleus in their cells (Ricaud et al., 1989).
Infective mycelia are capable to penetrate through buds at each sugarcane node and shortly reach apical meristem systemically. Infective buds in mature plants are either symptomatic as whip at end of stalk or asymptomatic hidden in buds up to next season when they are planted (Agnitori, 1990).
The microscopic detection of sugarcane smut fungus is not exact and quite difficult at early stages of plant colonization both in field and laboratory conditions Moosawi-Jorf and Izadi, 2007) because in the infected tissues smut hypha cannot be recognized morphologically from other fungal hypha.
Methods based on PCR molecular methods using specific primers were reported to detect specifically with high sensitivity the presence of small quantities of smut DNA mixed with sugarcane DNA (Schenck, 1998; Moosawi-Jorf and Izadi, 2007).
Teliospores of sugarcane smut in the whips of infected plants are surface infected with various infections including yeast bacteria and other saprophytic fungi that case some problems isolation, germination and purification of the teliospores.
The objective of this study was the isolation of different colonies of sugarcane smut from teliospore germination and from infected plants cultured on media and its identification in comparison with other infections using Polymerase Chain Reaction (PCR).
MATERIALS AND METHODS
Teliospores of sugarcane smut in the whips of infected plants are surface infected with various infections including yeast bacteria and other saprophytic fungi.
Isolation of yeast-like and mycelial colonies from teliospores: Whips of smutted plants of sugarcanes were collected from fields of Khuzestan province, Iran. Parts belonging 15 cm from tip and below of whips were cut and removed and then the middle part of whips were placed in pockets, labeled and transferred to laboratory. Whips were air dried for about 24 h under ambient temperature in laboratory, Sugarcane Research Institute, Ahwaz, Iran in 2005-2006. The whips containing teliospores were shaken and the waste materials were removed. Teliospores were transferred into sterile vials and incubated in sterile desiccators containing calcium chloride to reduce the humidity of teliospore mass. The teliospores were kept in air-tight containers frozen at 20°C for subsequent investigation.
Germinated teliospores: The teliospores were surface disinfected for 2 h. in sterile distilled water amended with 0.3 mg L-1 streptomycin sulfate. The teliospore suspension was streaked on slides using sterile loop. To each slide, PDA culture medium (containing streptomycin sulfate Fereol (1984)) was added. The slides were placed in plates and were incubated in the dark for 12 h at 28-30°C. The germinated teliospores were examined microscopically. They were then transferred by sterile needle into plates containing fresh medium followed incubation in the dark for 24 h at 28-30°C.
The 24 h old cultures of germinated teliospores were spread on YGC medium and incubated in dark for 48 h at 28-30°C for the preparation and purification of the yeast-like colonies. The yeast-like monocolonies produced on the culture, were streaked again by loop on YGC and the incubation step was repeated as described earlier.
Isolation of dikaryotic mycelia from infected plant tissues: Pieces (2-5 cm) of the stems from the infected plant tissues were surface disinfected with 0.05% sodium hypochlorite, rinsed with sterile distilled water three times and were plated into the PDA medium (amended with 250 mg L-1 streptomycin sulfate) followed by incubation in the dark at 28-30°C. The rate of mycelia growth was monitored daily for later isolation of dikaryotic mycelia (Moosawi-Jorf et al., 2006).
The cytology of mycelium was carried out using the Acridin Orange stain (1 mg acridin orang /ml phosphate buffer, 1 mol L-1, pH = 6). The acridin orange stains the nuclei. Therefore, mycelial mass were firstly immersed in 70% acetic acid for 1 min and then mounted in stain of acridin orange. The mycelial were examined microscopically using a microscope equipped with epifluorescent condenser and fluorescence cube of WB (490-500 nm excitation wavelength) (Olympus model BX51) (Alizadeh et al., 2001).
Polymerase Chain Reaction (PCR) amplification: The DNA was extracted from yeast-like and mycelial colonies of sugarcane smut, Fusarium colonies and bacterial colonies that infected the surface of teliospores as positive check following the method of Murray and Thampson (1980).
PCR amplification was carried out in a total volume of 25 μL containing 2 mL of extracted DNA, 0.24 μM each of the specific primers bE4 (5’-CgCTCTggTTCATCAACg-3’) and bE8 (5’-TgCTgTCgATggAAggTgT-3’) which amplified the mating type gene of U. scitaminea (Albert and Schench, 1996). The reaction mixture also contained 0.2 mM dNTPs, 0.5 mM MgCl2 and 1.5 unit μL-1 Taq DNA polymerase.
The reaction was run for 30 cycles of denaturation at 94°C for 30 s, annealing at 57°C for 30 s and extension at 72°C for 1 min. Finally, 7 μL of each PCR product was analyzed by electrophoresis. Samples of 7 μL PCR products were mixed with 2 μL of 10X loading dye (0.25% bromo-phenol blue, 0.25% xylene cyanol and 40% sucrose, W/V) and spun briefly before loading (Shaif-Nabi et al., 2001). The amplification products were analyzed on 1.2% agarose gel in 1xTBE buffer running at 70 volts for 2 h staining in 0.5 μL mL-1 ethidium bromide for 10 min and visualized under UV light. The DNA size marker used was λDNA double digested with EcoRI and HindIII.
DNA from surface infected teliospores Fusarium colonies was as positive check; and sterile water was used as negative check.
RESULTS
Isolation and purification of yeast-like and mycelial colonies from teliospores: Upon culturing the teliospore on PDA without antibiotics, bacteria grew on the surface of all culture media after 24 h; whereas, the white mycelia mixed with bacterial colonies were seen 48 h after culturing.
Teliospores began to germinate after 20 min of culturing on PDA amended with streptomycin sulfate and YGC. Pre-immersion of teliospores in streptomycin sulfate for 2 h was free of bacterial infection on these media.
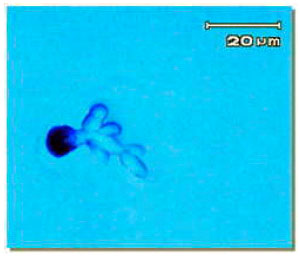 | |
| Fig. 1: | Teliospore germination of Ustilago scitaminea |
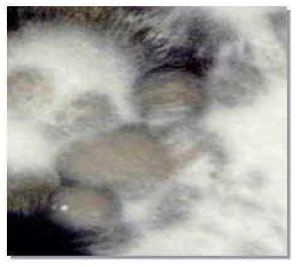 | |
| Fig. 2: | Yeast-like and mycelial colonies of Ustilago scitaminea |
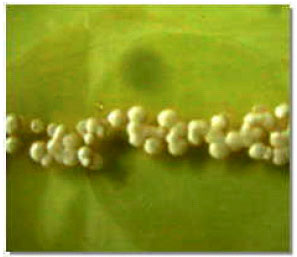 | |
| Fig. 3: | Yeast-like colonies of Ustilago scitaminea, produced monosporidia |
 | |
| Fig. 4: | White floccose erected mycelia of Ustilago scitaminea at the point where the (–) and (+) mating type were crossed on culture medium |
Teliospores germinated on YGC medium after 24 h and produced promycelia and sporidia (Fig. 1) or produced mycelia 48 h after spreading the germinated teliospores on YGC, yeast-like and white floccose erected mycelial colonies were produced as mixture colonies on medium (Fig. 2).
Monosporidial cultures were obtained by streaking of yeast-like colonies on YGC medium after 48 h at 28°C (Fig. 3). Sporidia of monosporidial cultures were multiplied in PS broth medium after 48 h at 28°C. Cross-streaking of positive and negative mating types of monosporidial sporidia were leaded to form the white floccose erected mycelial dikaryon at the point where they cross on culture medium after 48 h (Fig. 4).
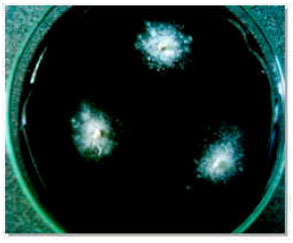 | |
| Fig. 5: | White mycelial growth of Ustilago scitaminea from infected pieces of sugarcane |
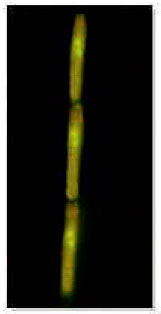 | |
| Fig. 6: | Dikaryotic hyphae of mycelial colonies of Ustilago scitaminea (Stained with Acridin Orange) |
Isolation of dikaryotic mycelia from smutted plant: White mycelial colonies grew on PDA, 5 days after culturing of pieces of stems from infected plants. White hyphae were uniformly spread on and around all cultured pieces (Fig. 5). Staining of nuclei of the mycelial colonies revealed the existence of two separated nucleuses in each cell of mycelia (Fig. 6).
Detection of the fungus by PCR
Smut identification by PCR in yeast-like and mycelial colonies: To detect the presence of the smut in samples of different treatment by PCR; specific primers bE4 and bE8 were used. The primers specifically amplified and detected a DNA fragment of 459 bp at annealing temperature of 57°C in all the treatments of yeast-like and dikaryotic forms.
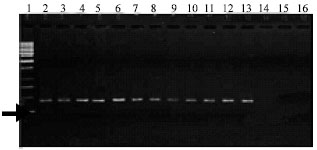 | |
| Fig. 7: | Amplification of 459 bp fragment from sugarcane smut in DNA extracted from sporidial yeast-like colonies. (1: Marker; 2-14: DNA extracted from Sporidia; 14: DNA extracted from yeast-bacterial colonies; 15: DNA extracted from Fusarium; 16: sterile distilled water) |
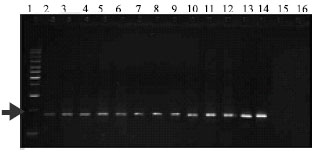 | |
| Fig. 8: | Amplification of 459 bp fragment from sugarcane smut in DNA extracted from dikaryotic mycelial colonies. (1: Marker; 2-7: DNA extracted from dikaryotic mycelia initiated from a mixture of + and – sporidia; 8-14: DNA extracted from dikaryotic mycelia initiated from infected plants; 15: DNA extracted from Fusarium; 16: Sterile distilled water) |
This DNA fragment was not amplified in DNA extraction of yeast-bacterial colonies and other saprophytic-mycelial fungi as well as in pure water as check specimens.
The PCR assay for DNA extracted from sporidia of yeast-like colonies, specifically yielded a positive response to amplify primers bE4 and bE8 (Fig. 7).
The PCR assay for DNA extracted from dikaryotic mycelia initiated from a mixture of + and-sporidia and/or from infected plant tissue plants, specifically yielded a positive response to amplify primers bE4 and bE8 (Fig. 8).
DNA extracted from Fusarium and sterile distilled water did not yield any amplification products under similar PCR conditions. Whereas, primers bE4 and bE8 of U. scitaminea specifically amplified a DNA fragment of 459 bp at an annealing temperature of 57°C in all the treatments of the DNA extracted from sporidial and mycelial cultures in vitro.
DISCUSSION
This technique of Albert and Schenck (1996) for disinfection was modified slightly to disinfect teliospore surfaces in streptomycin sulfate suspension for 2 h This disinfection treatment resulted in the deposition of all teliospores in the antibiotic solution and no bacterial infection on culture media was seen. This method enabled the control of abundant bacterial contamination, which is problematic in isolation and purification of the smut fungus. Present findings confirm the results reported earlier by Albert and Schenck (1996).
Using PDA amended with streptomycin sulfate and YGC containing chloramfenicol as media in this study for culturing of teliospores lead to full prohibition of bacterial contamination in the culture media. However, fungal Fusarium contamination was not controlled in this media, so isolation of the smut colonies from this fungal colonies was difficult. Sinha and Singh (1983) reported that teliospore germination of U. scitaminea is highly reduced in existence of Fusarium moniliforme var. subglatinans.
Fereol (1984) isolated and purified yeast-like and mycelial colonies of sugarcane smut successfully by the use of PDA amended with streptomycin sulfate. The incorporation of yeast extract and the glucose mono sugar in YGC, accelerated the germination and colony growth of the smut fungus, thus enabling the isolation and sub culturing of yeast-like colonies of the smut before development of probable saprophytic fungi. Pross and Chagrardieff (1983) examined many different sugars in culture media as carbon source for growth of sugarcane smut and they reported that maximum growth was obtained when glucose was the carbon source.
In this study, white hypha were uniformly isolated from infected plants below the whips. Two separated nuclei existed in each cell of mycelia upon staining with acridin orange. Ricaud et al. (1989) isolated three kinds of colonies of U. scitaminea including yeast-like, white mycelial and black mycelial colonies. Based on the present findings and the earlier reported results of Ricaud et al. (1989) it can be concluded that the sugarcane smut exist in dikaryotic mycelial form in infected plants including whips. Dikaryotization between two different mating types of sporidia in this study was confirmed, because mycelium was produced on media due to culturing of two different mating types of sporidia. Present results agree with those of Agnitori (1990), who suggested that two germ tubes of different mating types of sporidia can fuse to produce dikaryotic mycelia.
The specific primers bE4 and bE8 were used in the present study to detect smut fungal colonies initiated from teliospore germination and smutted sugarcane plants. The PCR assay detected smut DNA in all kinds of sugarcane smut colonies. Albert and Schenck (1996) amplified a homolog of bE mating-type gene of sugarcane smut using PCR assay. These primers sensitively detected the presence of U. scitaminea DNA. Other investigators used those specific primers to detect sugarcane smut DNA failed to detect the smut microscopically even one week after inoculation (Singh et al., 2004).
The present results revealed that using specific primers PCR assay can exactly and quickly identify and detect the yeast-like and mycelial colonies of sugarcane smut. Furthermore, this method differentiated smut colonies from other microorganisms including bacteria and saprophytic fungi that exist along with collected teliospores. The high specificity of this assay, detection of these colonies in infected plant tissues is possible (Moosawi-Jorf and Izadi, 2007).
REFERENCES
- Albert, H.H. and S. Schenck, 1996. PCR amplification from a homolog of the bE mating type gene as a sensitive assay for the presence of Ustilago scitaminea DNA. Plant Dis., 80: 1189-1192.
Direct Link - Moosawi-Jorf, S.A. and M.B. Izadi, 2007. In vitro detection of yeast-like and mycelial colonies of Ustilago scitaminea in tissue-cultured plantlets of sugarcane using polymerase chain reaction. J. Applied Sci., 7: 3768-3773.
CrossRefDirect Link - Murray, M.G. and W.F. Thompson, 1980. Rapid isolation of high molecular weight plant DNA. Nucl. Acids Res., 8: 4321-4326.
CrossRefPubMedDirect Link - Shaif-Nabi, B., D.K. Agarwal and N. Mitter, 2001. Differentiation of isolates of Neovossia indica by RAPD-PCR and clustering based on teliospore morphology. Acta Phytopathol. Entomol. Hungar., 36: 223-236.
Direct Link - Singh, N., B.M. Somai and D. Pillay, 2004. Smut disease assessment by PCR and microscopy in inoculated tissue cultured sugarcane cultivar. Plant Sci., 167: 987-994.
CrossRef