
Muhummadh Khan
Department of Bioinformatics, Research Wing, Indo American Cancer Hospital and Research Center, Hyderabad, India
Kaiser Jamil
Department of Bioinformatics, Research Wing, Indo American Cancer Hospital and Research Center, Hyderabad, India
Trends in Bioinformatics
Year: 2008 | Volume: 1 | Issue: 1 | Page No.: 7-17
DOI: 10.3923/tb.2008.7.17







ABSTRACT
This study determines conservation and functional divergence of Methylenetetrahydrofolate reductase (MTHFR) focusing on sites which bear single nucleotide polymorphisms (SNPs). We constructed its phylogeny and calculated type 1 divergence to identify regions of MTHFR under selection. We found that MTHFR is present throughout the three domains of life; Archaea, Bacteria and Eukarya. MTHFR well conserved preserving its function. It has been reported in earlier studies that polymorphisms in this gene are potential modulators of therapeutic response to anticancer drugs. Hence, SNPs in protein coding region were analyzed for the conservation and functional constraints. We report in this study that SNPs of MTHFR gene occur at sites under functional constraint i.e., these sites tend to be conserved. It is possible that SNPs at such sites could be fixed and propagated in a given population.
PDF Abstract XML References Citation
How to cite this article
Muhummadh Khan and Kaiser Jamil, 2008. Study on the Conserved and the Polymorphic Sites of MTHFR Using Bioinformatics Approaches. Trends in Bioinformatics, 1: 7-17.
DOI: 10.3923/tb.2008.7.17
URL: https://scialert.net/abstract/?doi=tb.2008.7.17
DOI: 10.3923/tb.2008.7.17
URL: https://scialert.net/abstract/?doi=tb.2008.7.17
INTRODUCTION
One-carbon folate metabolism is a network of interrelated biochemical reactions in which one-carbon unit is transferred to tetrahydrofolate for subsequent reduction or oxidation. The transfer of this one-carbon moiety is essential for DNA synthesis and biological methylation. Hence, folate is critically important to the viability of proliferating cells (Assaraf, 2007). Methylenetetrahydrofolate reductase (MTHFR) is a key enzyme in the folate metabolism. It is a flavoprotein and a member of the MTHFR family (EC 1.5.1.20). It catalyzes the irreversible reduction of 5, 10-methylenetetrahydrofolate to 5-methyltetrahydrofolate, thus generating the active and primary circulating form of folate required for remethylation of homocysteine to methionine. The human MTHFR gene is located on chromosome 1p36.3. The entire coding region has a length of 1.980 bp with a predicted molecular mass of 74.6 kDa (Frosst et al., 1995; Goyette et al., 1998). MTHFR contains 11 exons with a length ranging from 102 to 432 bp and introns ranging from 250 bp to 1.5 kb, except for one intron of 4.2 kb. So far, two mRNAs of 7.5 and 8.5 kb have been identified in tissues. Several alternatively spliced 5` non-coding regions have been reported, possibly indicating complexity of the MTHFR gene expression (Goyette et al., 1998).
Two common single nucleotide polymorphisms (SNP) have been described to affect the activity of the MTHFR enzyme. The first SNP to be reported in MTHFR was the c677t variant (Frosst et al., 1995). This SNP occurs in exon 4 and results in an alanine to valine substitution at codon 222. The polymorphism lies in the binding site for the MTHFR co-factor flavin adenine dinucleotide (FAD). Individuals with MTHFR C677T TT genotype have been shown to have 30% in vitro MTHFR enzyme activity as compared with the wild type, whereas those with the heterozygous (CT) genotype have been found to have 60% of wild type MTHFR enzyme activity (Goyette et al., 1998). The disruption of homocysteine metabolism by this polymorphism influences risk for several complex disorders, including cardiovascular disease, neural tube defects and some cancers. A second MTHFR polymorphism, A1298C in exon 7, results in a glutamate to alanine substitution at codon 429 (Van Der Put et al., 1998; Weisberg et al., 1998). This polymorphism lies in the S-adenosylmethionine-regulatory domain of the enzyme (Matthews et al., 1984; Jencks and Mathews, 1987). The binding of S-adenosylmethionine (SAM) results in conformational changes within the MTHFR enzyme which inhibit the enzyme`s activity (Jencks and Mathews, 1987).
Though, MTHFR is not an oncogene, SNPs in MTHFR are potential modulators of response to anticancer drugs. Several studies have investigated MTHFR gene variants, disease and disorders involving folate metabolism (Nakata et al., 1998; Robien and Ulrich, 2003; Schwahn and Rozen, 2001). It was reported in earlier studies that there was an increased risk of breast cancer and acute lymphoblastic leukemia due to the two common SNPs of MTHFR in the South Indian population (Reddy and Jamil, 2006; Kalyankumar and Jamil, 2006).
The objective of the present study was to determine evolutionary history, species spread and conservation of this enzyme. Site specific functional divergence was estimated in order to identify regions of MTHFR undergoing purifying selection. Occurrence of a SNP in the genome is at random. In many association studies, SNPs are reported to occur in a given population consistently. Hence these are used as ideal biomarkers (Reddy and Jamil, 2006; Kalyankumar and Jamil, 2006). We were interested to know whether the functional pressure, at a given amino acid site in a given protein, aids in fixing and transmission of SNPs in a given population. Hence the level of functional constraint at sites where the SNPs have occurred in MTHFR was noted. This was done to identify any pattern in constraints at these sites.
MATERIALS AND METHODS
This study involves information on MTHFR gene polymorphisms from our studies and those from other studies (Reddy and Jamil, 2006; Kalyankumar and Jamil, 2006). Using bioinformatics tools, as described in the methodology below, we have carried out this investigation in the bioinformatics lab of our institute.
Data Set and Sequence Alignment
The homologous protein sequences of human MTHFR were retrieved from the NCBI REFSEQ using protein BLAST (Altschul et al., 1997; Pruitt et al., 2007). The similarity search yielded many homologs. Hence, a representative dataset of 73 sequences was assembled from the resultant sequences. These sequences were chosen to represent the given class of organisms. The sequence similarity of homologs to the human sequence of MTHFR was more than 30%. These sequences were aligned using MUSCLE with default settings (Edgar, 2004). The resulting multiple alignment was manually edited using (Jalview Clamp et al., 2004). In the present study, this alignment was used as input for further analyses.
Phylogenetic Tree Building
Phylogenetic tree was generated by neighbour joining method implementing JTT substitution model with complete deletion of gaps. All substitutions were included with homogeneous pattern among lineages and uniform rates among sites. The statistical support was provided by generating 1000 bootstrap replicates. The tree was drawn, viewed and edited with tree explorer. All of phylogenetic reconstruction was done using MEGA 3.0 (Kumar et al., 2004).
Functional Divergence and Stability of the Coding SNPs
Functional divergence between bacterial and eukaryotic clusters, fungi and animalia clusters and amongst major bacterial classes was calculated using DIVERGE 1.04 (Gu and Elden, 2002). The sites bearing the SNPs were also included and the functional constraint was noted. The sites estimated to have functional divergence greater then 0.6 were plotted onto the three dimensional structure of Thermus thermophilus (PDB id: 1v93). The graphic manipulations to the protein structure were done using PYMOL DeLano (2006).
RESULTS AND DISCUSSION
Cancer cells show hyper folate activity which made this metabolic pathway a potential target for anticancer drugs. Of late, MTHFR, which is a key enzyme in the pathway, has been the subject of many investigations. This is due to the fact that SNPs in this gene modulate therapeutic response to anticancer drugs. Investigators have hypothesized that polymorphisms in the MTHFR may result in differential toxicity to cyclophosphamide, methotrexate and fluorouracil (Toffoli et al., 2000). A vivid understanding of the structural properties of MTHFR is needed to understand its role in elevated toxicity to chemotherapeutics, increased risk to breast cancer and reduced risk to lung and colon cancer. In the present study, We performed the phylogenetic analysis of MTHFR in order to understand the evolution and conservation of this enzyme. We also estimated its functional divergence and basing on this we attempted to predict the characteristics of the site at which there is high probability for an SNP to occur.
Phylogenetic Tree of MTHFR
The phylogenetic tree was constructed using homologs belonging to diverse species ranging from Archaea to Bacteria to Eukarya (Fig. 1). The blast search yielded more than 500 sequences. Hence representative sequences were chosen from major classes of organisms to constitute a final dataset of 73 sequences. Though the sequence homologs obtained by similarity search showed bacterial sequences to be at least 200 residues shorter than the eukaryotic counter parts, the alignment of the MTHFR domains leaves no doubt that the structure is conserved. It is clear from our multiple alignment (Fig. 2) that the active catalytic residues aspartic acid and glutamic acid are conserved in all the representative sequences from the three domains of life. Even with such diversification in species, MTHFR still retains its function and structure.
The pathway of one carbon folate seems to be ancient, since it caters to the fundamental requirement of the nucleotide synthesis and control of gene expression. The reaction catalyzed by MTHFR is irreversible. This acts as a one way valve by blocking the dissolution of the folate pool. This gateway function of MTHFR could be assumed to be as ancient as the pathway itself, since the flow of products in one carbon folate pathway is essential for the maintenance of folate pool which in turn is important raw material sanctuary for various cellular processes as stated earlier.
Large variety of bacterial species dominate the inferred phylogenetic tree (Fig. 1). All the major classes of the bacterial kingdom have the MTHFR gene. During the tree building process, large genetic distances were encountered between Archaean and Bacterial sequences indicating the differential MTHFR evolution in the two domains. Though domain structure and enzyme function of bacterial and eukaryotic MTHFR is the same, phylogenetic tree building clearly groups the homologs into two distinct clusters. The Eukaryotic subtree is clustered into two clades. The homologs of animalia species were clustered as one and the other contained many fungi along with plants and other unicellular eukaryotes. This clustering of homologs demarks the different paths evolution that the ancestral MTHFR has undergone.
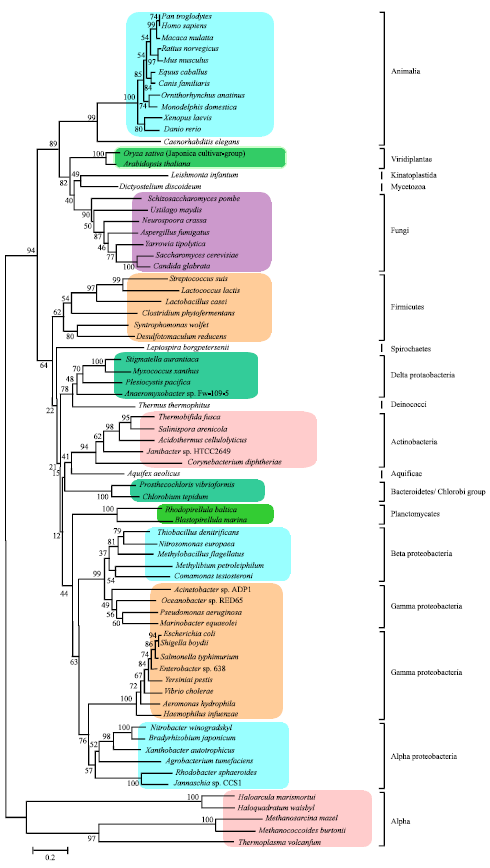 | |
| Fig. 1: | Phylogenetic tree of MTHFR |
 | |
| Fig. 2: | The conserved active residue sites 103 and 199 are marked |
Functional Divergence
Functional divergence identifies amino sites that have high probability to diverge from conservation. These sites are assumed to be undergoing purifying selection. The protein products of two functionally different but closely related genes show high probability of functional divergence at those sites which are responsible for the change in function. In the present study we identified the different residues undergoing active evolution are different in eukaryotes and the prokaryotes.
Functional divergence of a protein could occur after major evolutionary events such as gene duplication or speciation. Some of them may result in altered selective constraints (different evolutionary rates) at certain amino acid residues, which are called type 1 functional divergence, regardless of the underlying evolutionary mechanisms (Kutzbach and Stokstad, 1971). In the present study, Phylogenetic tree of MTHFR was grouped into different clusters. These selected clusters were taken into consideration for calculating the type I divergence. Functional divergence was calculated for different sets compromising different selected clusters.
In this study the site specific divergence θ was calculated for the clusters of a set individually. For selected clusters, if θ = 0 then it implies that there is no site specific divergence. If the θ > 0 then it means that this particular site in the protein has undergoing purifying selection. Given that the θ estimations are biased by alignment error, care was taken to have an accurate multiple alignment. Detection of site-specific rate shifts can provide a list of predicted amino acid residues that may be responsible for functional divergence between member genes of a gene family (Gu, 2001). The reader is referred to some of the studies that support the concept of functional divergence for further information (Gaucher et al., 2001; Jordan et al., 2001).
Early molecular evolutionary studies have unequivocally shown that different genes evolve at substantially different rates. Type I divergence was calculated to observe the functional constraints on proteins which limit their evolutionary rates at specific sites. These constraints allow for the interpretation of sites of conserved residues and sites with a rate change as those most likely underlying the functional similarities and differences among protein subfamilies. The height of each bar in the graph represents the posterior probability of any given site as labeled on the x-axis (Fig. 3a-c). The significance of these values is that a low value denotes high constraint and vice versa. This indicates the extent of divergence of amino acid residues in any given protein.
The first set consisted of eukaryotic and bacterial clusters (Fig. 3a). There are 254 sites in all showing discernable rates of divergence. A stringent cut off of 0.7 was employed to get the prominent sites where the divergence is high. There are 23 such sites. This indicates that at these sites evolutionary rate is high in eukaryotes in comparison with bacteria. These were plotted onto the structure of protein as spheres of red colour (Fig. 3d). There were about 36 sites, seen as spheres of green colour as seen in figure, which showed negligible divergence when the cut off value of 0.1 was employed. These sites indicate slower rate of evolution in eukaryotes at these residues in comparison with bacteria. The active residues showed negligible values indicating high levels of functional constraint.
The second set consists of fungi-plants and animalia clusters (Fig. 3b). The pairwise comparison yielded 24 sites out of 254 showing high divergence when a cutoff of 0.7 was employed. The active residues show less value when compared to rest of 254 sites. The third set consisted of major bacterial classes as individual clusters; Firmicutes, Actinobacteria, Delta-Epsilon group and Proteobacteria. These clusters were cross compared with each other. The results were plotted onto a single graph (Fig. 3c). When cut off of 0.7 was employed, about 27 sites showed high divergence values in all the comparisons. Amongst the comparisons the least divergence was estimated between Firmicutes and Delta-Epsilon clusters. The highest was between Actinobacteria and Proteobacteria clusters. This indicates different rates of evolution amongst the major bacterial classes. The sites showing
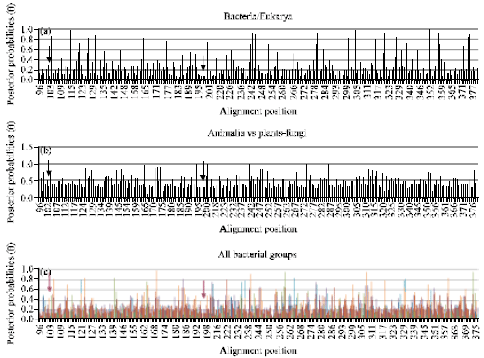 | |
| Fig. 3a-c: | Graphs showing the posterior probabilities at various sites for the (a) Bacteria vs. Eukarya, (b) Animalia vs. Plants-Fungi and (c) all bacterial groups. The Y-axis shows the posterior probabilities and X-axis shows the sites on the alignment. The higher probability value indicates higher divergence at the given site. The active residues sites are 103 and 199 are indicated by the red arrows. These show negligible divergence indicating high functional constraint |
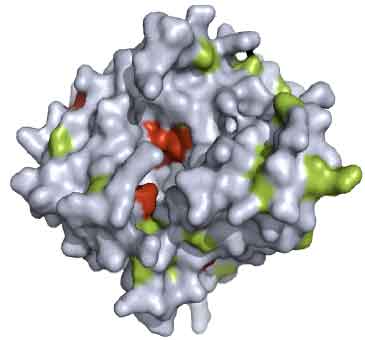 | |
| Fig. 3d: | The protein structure of MTHFR showing sites in Green which are evolving faster in eukaryote than in prokaryotes and sites in red which are evolving slower in eukaryote. Active sites are colored red. The green regions are dispersed on the surface and the red regions are mostly in the interior of the protein |
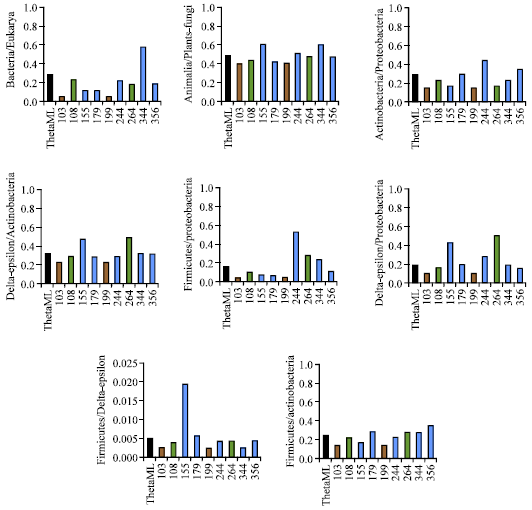 | |
| Fig. 3e: | Each graph represents the probability values for conserved and the SNP sites between two different clusters. The Y- axis shows posterior probability of specific sites and the X-axis denoting the alignment position. The Theta ML for the group is given basing on which the conserved and SNP sites` probabilities are interpreted. Site 103 and 199 are the conserved sites (red). The probability values are very less indicating functional constraint. Sites 108 and 264 are the nonsynonymous SNPs sites (green). The remaining are the synonymous sites (blue). In general SNPs posterior probabilities are near to the groups` Theta ML. The site 264 is less or very near to the ThetaML in most of the comparisons except the Delta-Epsilon/Proteobacteria graph. The Firmicutes/Delta-Epsilon cluster comparison shows negligible values for all the sites hence the Y-axis of the graph is rescaled with values ranging from 0 to 0.025 |
probabilities of more than 0.9 are different in eukaryotes and bacteria. This shows that regions undergoing functional divergence in Eukaryotes and bacteria are distinct and different. It is to be noted that even in this graph the active residues at sites 103 and 199 show negligible divergence.
Functional Constraints on SNP Bearing Sites in Coding Region of MTHFR
A SNP is a polymorphic base where the point mutation has persisted in a given population. MTHFR has been under the limelight of research owing to its SNPs C677T and A1298C. Both of these have been implicated in increased and decreased risks of various disorders. The extent, nature and magnitude of selection on amino acid variants are also relevant to understanding the relation between human disease and genetic variation (Wright et al., 2003; Burger et al., 2006). Bearing this we analyzed conservation and mutability of sites at which the SNPs of MTHFR occur.
Mutations are known to occur at different rates in exonic, intronic and intergenic regions of a genome. It is well established that nonsynonymous sites in protein-coding sequences are subject to purifying selection caused by constraints operating at the level of protein structure and function and that positive selection that, at least in mammals, affects a minority of genes and/or sites is an important force of adaptive evolution (Sunyaev et al., 2001; Wright et al., 2003; Bustamante et al., 2005). To explore the properties of the SNPs, which are nothing but persistent mutations, we analyzed the seven sites in MTHFR where the SNPs are found. We included the five sites which showed the synonymous substitutions because it has been recently reported that positive selection is wide spread in even the synonymous sites (Nielsen et al., 2005). When the SNP sites have to be analyzed for conservation or divergence from such a large variety of species, it could be speculated that this procedure might yield confusing results. But basing on assumption that in contrast to genotypic mutation rates, there does not seem to be a significant difference between eukaryotic and prokaryotic phenotypic mutation rates (Vallender and Lahn, 2004) we can expect that the mutation rates in MTHFR to be nearly the same in all species represented in the phylogenetic tree. In our estimation of functional divergence the sites with SNPs site showed less divergence probabilities (Fig. 3e). Since SNPs are the result of nucleotide substitutions, this finding was contrary to the assumption that SNP sites would show good divergent probabilities. All the seven SNP sites in general showed less divergent probabilities in comparison to the conserved sites and the ThetaML of the group verified (Fig. 3e). The conservation scores for nonsynonymous were a little higher than the conserved sites indicating that these sites have been under certain level of evolutionary constraint.
All nucleotides in the DNA are under different levels of functional constraints. The constraints are greater for the protein coding genes. The nucleotide sites which are under the highest constraint are those which correspond to the conserved sites in the protein (example, catalytic sites). The sites which are under less or no functional constraint are highly divergent and are subjected to positive selection. We found that the SNPs of MTHFR are under certain level of constraint. But the evolutionary pressure is not as high as at a conserved site and not as less as a site undergoing positive selection. This would imply that mutations at these sites would occur at a slow rate due to evolutionary constraints. Hence gaining a mutation at this site would be rare but nonetheless possible in higher frequency than at a conserved site. Owing to the evolutionary pressure at such a site, gained mutation is retained long enough with less chance of a reversion or gaining a forward mutation. This provides ample time for the mutation to be fixed in the gene. If this happens in germ cells then it could be inherited. This type of site is most probable position for stable SNP to occur. The allelic frequency of SNP variant genes might depend on this criteria; If in a given population, a SNP has occurred at such a site and the population is known to be reproducing without external hindrances, then this SNP would be widespread and stable in that population. The highest frequency allele would bear this SNP owing to the stability of the site.
CONCLUSION
MTHFR is an important enzyme owing to its role in the folate metabolism. It is present in diverse species from the three domains of life; Archaea, Bacteria and Eukarya. We found that the sites showing significant divergence were different for Bacteria and Eukarya; indicating at the variant evolutionary trends of MTHFR in these organisms. It is concluded that the SNPs of MTHFR occur in sites which are under functional constraint, but this constraint is less in comparison with conserved sites and more in comparison with a variable sites of the same protein. Hence, we speculate that this could be a possible reason for the fixing of SNPs in MTHFR as it provides a statistical reasoning behind the occurrence of SNPs in protein coding genes. From this study we presume that the allelic frequencies could be based on functional constraints at the site of SNP. But the contribution of the fixed SNP to the structural destabilization of a given protein is subjected to stochastic processes. Simply put, it should occur at the right place at the right time.
ACKNOWLEDGMENTS
We are grateful to the Director, Dr. Mohana Ch. Vamsy and Scientific Advisers of Indo American Cancer Institute and Research Centre for their encouragement and support. We are thankful to the Chairman and Dean and MGNIRSA for their support.
REFERENCES
- Altschul, S.F., T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller and D.J. Lipman, 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. Acids Res., 25: 3389-3402.
CrossRefPubMedDirect Link - Assaraf, Y.G., 2007. Molecular basis of antifolate resistance. Cancer Metastasis Rev., 26: 153-181.
Direct Link - Burger, R., M. Willensdorfer and M.A. Nowak, 2006. Why are phenotypic mutation rates much higher than genotypic mutation rates? Genetics, 172: 197-206.
Direct Link - Bustamante, C.D., A. Fledel-Alon, S. Williamson, R. Nielsen and M.T. Hubisz et al., 2005. Natural selection on protein-coding genes in the human genome. Nature, 437: 1153-1157.
Direct Link - Clamp, M., J. Cuff, S.M. Searle and G.J. Barton, 2004. The Jalview Java alignment editor. Bioinformatics, 20: 426-427.
Direct Link - Edgar, R.C., 2004. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res., 32: 1792-1797.
CrossRefDirect Link - Frosst, P., H.J. Blom, R. Milos, P. Goyette and C.A. Sheppard et al., 1995. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet., 10: 111-113.
CrossRefDirect Link - Gaucher, E.A., M.M. Miyamoto and S.A. Benner, 2001. Function-structure analysis of proteins using covarion-based evolutionary approaches: Elongation factors. Proc. Natl. Acad Sci. USA., 98: 548-552.
Direct Link - Goyette, P., A. Pai, R. Milos, P. Frosst and P. Tran et al., 1998. Gene structure of human and mouse methylenetetrahydrofolate reductase (MTHFR). Mamm Genome, 9: 652-656.
CrossRefDirect Link - Gu, X., 2001. Maximum-likelihood approach for gene family evolution under functional divergence. Mol. Biol. Evol., 18: 453-464.
Direct Link - Gu, X. and K.V. Elden, 2002. Velden, DIVERGE: Phylogeny-based analysis for functional-structural divergence of a protein family. Bioinformatics, 18: 500-501.
Direct Link - Jencks, D.A. and R.G. Mathews, 1987. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine. Effects of adenosylmethionine and NADPH on the equilibrium between active and inactive forms of the enzyme and on the kinetics of approach to equilibrium. J. Biol. Chem., 262: 2485-2493.
PubMedDirect Link - Jordan, I.K., G.R. Bishop and D.S. Gonzalez, 2001. Sequence and structural aspects of functional diversification in class I alpha-mannosidase evolution. Bioinformatics, 17: 965-976.
Direct Link - Kalyankumar, C. and K. Jamil, 2006. Methylenetetrahydrofolate reductase (MTHFR) C677T and A1298C polymorphisms and breast cancer in South Indian population. Int. J. Can. Res., 2: 143-151.
Direct Link - Kumar, S., K. Tamura and M. Nei, 2004. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform., 5: 150-163.
CrossRefPubMedDirect Link - Kutzbach, C. and E.L.r. Stokstad, 1971. Mammalian methylenetetrahydrofolate reductase Partial purification, properties and inhibition by S-adenosylmethionine. Biochim. Biophys. Acta (BBA)-Enzymol., 250: 459-477.
CrossRefDirect Link - Matthews, R.G., M.A. Vanoni, J.F. Hainfeld and J. Wall, 1984. Methylenetetrahydrofolate reductase. Evidence for spatially distinct subunit domains obtained by scanning transmission electron microscopy and limited proteolysis. J. Biol. Chem., 259: 11647-11650.
Direct Link - Nakata, Y., T. Katsuya, S. Takami, N. Sato and Y. Fu et al., 1998. Methylenetetrahydrofolate reductase gene polymorphism: Relation to blood pressure and cerebrovascular disease. Am. J. Hypertens., 11: 1019-1023.
CrossRefDirect Link - Nielsen, R., C. Bustamante, A.G. Clark, S. Glanowski and T.B. Sackton et al., 2005. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS. Biol., 3: e170-e170.
Direct Link - Pruitt, K.D., T. Tatusova and D.R. Maglott, 2007. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res., 35: D61-D65.
Direct Link - Reddy, H. and K. Jamil, 2006. Polymorphisms in the MTHFR gene and their possible association with susceptibility to childhood acute lymphocytic leukemia in an Indian population. Leukemia Lymphoma, 47: 1333-1339.
Direct Link - Robien, K. and C.M. Ulrich, 2003. 5,10-Methylenetetrahydrofolate reductase polymorphisms and leukemia risk: A HuGE minireview. Am. J. Epidemiol., 157: 571-582.
Direct Link - Schwahn, B. and R. Rozen, 2001. Polymorphisms in the methylenetetrahydrofolate reductase gene: Clinical consequences. Am. J. Pharmacogenomics, 1: 189-201.
CrossRefDirect Link - Sunyaev, S., V. Ramensky, I. Koch, W. Lathe, A.S. Kondrashov and P. Bork, 2001. Prediction of deleterious human alleles. Hum. Mol. Genet., 10: 591-597.
Direct Link - Toffoli, G., A. Veronesi, M. Boiocchi and D. Crivellari, 2000. MTHFR gene polymorphism and severe toxicity during adjuvant treatment of early breast cancer with cyclophosphamide, methotrexate and fluorouracil (CMF). Ann. Oncol., 11: 373-374.
Direct Link - Vallender, E.J. and B.T. Lahn, 2004. Positive selection on the human genome. Hum. Mol. Genet., 13: R245-R254.
Direct Link - Weisberg, I., P. Tran, B. Christensen, S. Sibani and R. Rozen, 1998. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol. Genet. Metab., 64: 169-172.
CrossRefDirect Link - Wright, A., B. Charlesworth, I. Rudan, A. Carothers and H. Campbell, 2003. A polygenic basis for late-onset disease. Trends Genet., 19: 97-106.
Direct Link - Van der Put, N.M., F. Gabreels, E.M. Stevens, J.A. Smeitink and F.J. Trijbels et al., 1998. A second common mutation in the methylenetetrahydrofolate reductase gene: An additional risk factor for neural-tube defects? Am. J. Hum. Genet., 62: 1044-1051.
PubMed