
Abdulaziz Ali A. Al-Khedhairy
Department of Zoology, College of Sciences, P. O. Box 2455
King Saud University, Riyadh 11451 Saudi Arabia
Misbahul Arfin
Department of Zoology, College of Sciences, P. O. Box 2455
King Saud University, Riyadh 11451 Saudi Arabia
Bassam A. Alahmadi
Department of Zoology, College of Sciences, P. O. Box 2455
King Saud University, Riyadh 11451 Saudi Arabia
Mohamed A. Al-Jumah
King Fahad National Guard Hospital, Riyadh, Saudi Arabia
Journal of Medical Sciences
Year: 2005 | Volume: 5 | Issue: 4 | Page No.: 275-279







ABSTRACT
Single Nucleotide Polymorphisms (SNPs) in presenilin-1 gene (PS1) were characterized in Saudi patients afflicted with Alzheimer’s Disease (AD) using Polymerase Chain Reaction (PCR) and automated DNA sequencing methods. Genomic DNA was extracted from blood of both patients and normal individuals using organic extraction methods. Genomic DNA was amplified for all PS1 exons and intronic boundaries. Fragments were sequenced and compared with the sequences of the respective exons of normal individuals as well as the data available in GenBank. Comparison yielded the discovery of two SNPs, T56240G and T56454C in 5′ and 3′ intronic regions of exon 8 of late-onset AD Saudi patients, respectively.
PDF Abstract XML References Citation
How to cite this article
Abdulaziz Ali A. Al-Khedhairy, Misbahul Arfin, Bassam A. Alahmadi and Mohamed A. Al-Jumah, 2005. Single Nucleotide Polymorphism Associated with Late-onset Alzheimer’s Disease. Journal of Medical Sciences, 5: 275-279.
DOI: 10.3923/jms.2005.275.279
URL: https://scialert.net/abstract/?doi=jms.2005.275.279
DOI: 10.3923/jms.2005.275.279
URL: https://scialert.net/abstract/?doi=jms.2005.275.279
INTRODUCTION
Alzheimer Disease (AD) is a neurodegenerative disorder and the most common cause of dementia in the elderly. Although the age of onset varies, most of the cases of AD occur at a late age (late-onset AD). Recently, considerable progress has been made in unraveling the complex etiology of AD. Four loci: amyloid precursor protein (APP), apolipoprotein E (ApoE), presenilin-1 (PS1) and presenilin-2 (PS2) genes have been identified contributing to AD[1-8].
E4 allele of ApoE locus on chromosome 19 has been associated with both familial and sporadic late-onset AD[9,10]. However, only half of all patients with AD have E4 allele and a substantial number of people having E4 allele do not develop AD. These facts indicate that another gene may also be involved particularly in sporadic AD. Further, Wragg et al.[11] reported an association between PS1 polymorphism and late-onset sporadic AD. PS1 has also been suggested as a potential risk factor for late-onset AD[12]. However, some other studied have shown conflicting reports[13].
Therefore, a study was carried out to search for Single Nucleotide Polymorphism (SNP) in PS1 in a number of Saudi AD patients. In earlier research, we reported the absence of mutations in PS1 of Saudi AD patients with late-onset AD[14]. This study describes the association of SNPs with late-onset AD in patients referred for diagnostic testing at King Fahad National Guard Hospital and King Khalid University Hospital, Riyadh, Saudi Arabia over a period of eight years.
MATERIALS AND METHODS
Saudi patients with Late-Onset Alzheimer’s Disease (LOAD) were recruited from those who visited King Fahad National Guard Hospital and King Khalid University Hospital during 1994-2001. All patients in the study had a probable diagnosis of AD according to DSM IV criteria[15] and were found to have AD at more than 65 years of age indicating the late-onset form of AD. The age of the patients at the time of testing ranged from 67 to 80 years with a mean age of 70 years. All the patients underwent detailed clinical evaluation, neuropsycological testing, CT Scan or MRI imaging of the brain and electroencephalography (EEG). Metabolic disorder and electrolytes abnormalities were excluded by the appropriate/specific tests. A number of age and sex- matched normal controls were also recruited from the same population.
DNA was extracted from the blood with standard procedures utilizing proteinase-K/phenol/chloroform extraction. Primers were designed on the basis of the sequence data for various exons of PS1 available in the GenBank to amplify the coding sequence of respective exons and flanking intronic regions. PCR was performed using PuRe Taq Ready-To-Go PCR Beads (Amersham Biosciences, USA) with different sets of primers (Table 1).
A 200 to 300 ng of genomic DNA was used as a template in 25 μL reaction. Genomic DNA was amplified for 40 cycles. Each cycle consisted of 94°C for 30 sec, 53°C for 30 sec, 72°C for 1 min.
PCR products obtained were separated by electrophoresis on 1.5% agarose gel in TAE buffer, visualized by ethidium bromide fluorescence. Fragments with the expected size were cut from the gel, purified using GFX PCR DNA Gel band purification kit (Amersham Biosciences, USA).
Purified DNA was sequenced using forward and/or backward primers. The DNA sequence was compared with PS1 genomic sequences from normal individuals and data available in Genbank.
RESULTS AND DISCUSSION
Amplified DNA when sequenced and compared with the sequences of the respective exon from normal and also with the data available in GenBank, showed two SNPs at the 5’ and 3’ intronic regions of exon 8 (encoding for amino acid 184 to 256). There were T56240G and T56454C in the 5’ and 3’ intronic region of exon 8, respectively (Fig. 1A and B).
| Table 1: | Set of primers used for various exons |
 | |
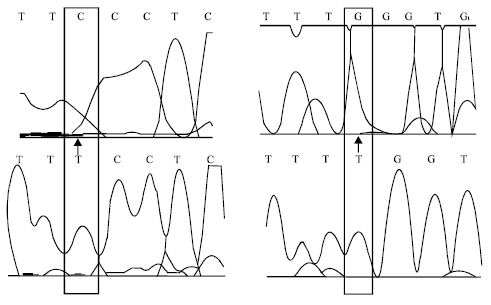 | |
| Fig. 1: | Comparison of electrophorograms of PS1 intronic DNA sequences of AD patients with those of normal human A. Showing the T56454C SNP. B. Showing the T56240G SNP |
These variations noticed in the intronic region seem to segregated with late-onset AD . No other variations were found in the coding as well as non coding sequences of exon 8 of all other AD patients, which was further confirmed by the comparison of their electrophorograms.
A search for SNPs in the PS1 gene in sporadic LOAD Saudi patients yielded the discovery of two SNPs. Furthermore, no mutations in PS1 coding sequences were found in sporadic LOAD patients. The absence of mutations in PS1 of LOAD patients is in agreement with the fact that these patients were over 65 yrs old. At this age group, as in the case of other ethnic group populations, most inherited AD-causing mutations are scattered in PS2 and ApoE specific genotype[12].
Moreover, PS1 mutations are linked with early-onset AD (EOAD) and more than 70 mutations have been reported in various exons of PS1 in different patients from various ethnic groups[16,17].
In present study, two SNPs were detected at two intronic regions within PS1 in sporadic LOAD patients. Intronic polymorphism in PS1 gene has also been reported in exon 8 of late-onset American AD patients[11]. Present results support the genetic association between SNPs in PS1 gene and LAOD as suggested by Wragg et al.[11].
The polymorphism found in PS1 of Saudi patients is biologically relevant to AD as we could not find any polymorphism in control subjects. Secondly, it may be in linkage dis-equilibrium with biologically relevant variability elsewhere in the PS1 gene. The position of the T56240G SNP in 5’ intronic region of exon 8 indicates that the most likely mode of action would be through the modulation of alternate splicing particularly of exon 7 and 8. The position of the T56454C SNP indicates that if the polymorphism is biologically relevant then the most likely mode of action would be through the modulation of the alternate splicing of exon 8 and 9 of PS1 gene.
Further, alternate splicing of exon 8 has been reported in some tissues[6] and a variation in the acceptor site in the same intron leads to early-onset disease through the loss of exon 9[18]. Furthemore, exon 8 is the site of most of the mutations leading to the early-onset AD (EOAD)[8,19]. However, in another related study, we could not find any mutation in exon 8 of EOAD Saudi patients (Al-Khedhairy et al. unpublished).
Various authors have reported some association of PS1 polymorphism with AD. Nishiwaki et al.[20] suggested that T/G SNP at intron 9 of PS1 gene is associated but not responsible for late-onset AD in Japanese. Yasuda et al.[13] reported a subtle but positive association of PS1 polymorphism with AD in Japanese population. Combarros et al.[21] examined the possible involvement of PS1 and PS2 polymorphism in the risk of sporadic AD and suggested that PS1 and PS2 polymorphism may increase the risk conferred by the presence of ApoE epsilon 4 allele.
Tang et al.[22] reported that PS1 polymorphism is only associated with EOAD but not with LOAD in Chinese. Similarly other workers reported no association or weak association of PS1 intronic polymorphism and LOAD in Chinese[23,24]. A -48 C/T polymorphism in PS1 promoter has been associated with an increased genetic risk in EOAD and has been shown to influence the expression of PS1 gene thereby influencing βA4 load[25].
Thus, a search for SNPs in PS1 in late-onset Saudi AD patients yielded the discovery of two SNPs (prevalence 3.4%) in two Saudi LOAD cases. Unfortunately, due to lack of information regarding family history, we could not follow the inheritance pattern. Finally, the cause of disease in the rest of the patients where no mutations/polymorphism can be detected is unknown and it may be result of certain mutations on other loci or other genetic and/or environmental factors as suggested by Axelman et al.[26].
ACKNOWLEDGMENT
A.A. Al-Khedhairy would like to thank King Abdulaziz City for Science and Technology for financial support (Project No. GP-18-1).
REFERENCES
- Goate, A., M.C. Chartier-Harlin, M. Mullan, J. Brown and F. Crawfor et al., 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature, 349: 704-706.
Direct Link - Pericak-Vance, M.A., J.L. Bebout, P.C. Gaskell, L.H. Yamaoka and W.Y. Hung et al., 1991. Linkage studies in familial Alzheimer disease: Evidence for chromosome 19 linkage. Am. J. Hum. Genet., 48: 1034-1050.
PubMedDirect Link - Mullan, M., F. Crawford, K. Axelman, H. Houlden, L. Lilius, B. Winblad and L. Lannfelt, 1992. Pathogenic mutations for probable Alzheimer's disease in the APP gene at N-terminus of beta-amyloid. Natl. Genet., 1: 345-347.
PubMedDirect Link - Saunders, A.M., W.J. Strittmatter, D. Schmechel, P.H. George-Hyslop and M.A. Pericak-Vance et al., 1993. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology, 43: 1467-1472.
Direct Link - Levy-Lahad, E., W. Wasco, P. Poorkaj, D.M. Romano and J. Oshima et al., 1995. Candidate gene for the chromosome familial Alzheimer's disease locus. Science, 269: 973-977.
PubMedDirect Link - Rogaev, E.I., R. Sherrington, E.A. Rogaeva, G. Levesque and M. Ikeda et al., 1995. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to Alzheimer's disease type3 gene. Nature, 376: 775-778.
PubMedDirect Link - Schellenberg, G.D., 1995. Genetic dissection of Alzheimer disease, a heterogeneous disorder. Proc. Natl. Acad. Sci. USA., 92: 8552-8559.
Direct Link - Sherrington, R., E.I. Rogaev, Y. Liang, E.A. Rogaeva and G. Levesque et al., 1995. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature, 375: 754-760.
PubMedDirect Link - Corder, E.H., A.M. Saunders, W.J. Strittmatter, D.E. Schmechel and P.C. Gaskell et al., 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science, 261: 921-923.
CrossRefDirect Link - Wragg, M., M. Hutton and C. Talbot, 1996. Genetic association between intronic polymorphism in presenilin-1 gene and late-onset Alzheimer's disease. Lancet, 347: 509-512.
Direct Link - Theuns, J., J. Del-Favero, B. Dermaut, C.M. van Duijn and H. Backhovens et al., 2000. Genetic variability in the regulatory region of presenilin 1 associated with risk for Alzheimer's disease and variable expression. Hum. Mol. Genet., 9: 325-331.
Direct Link - Tandon, A., E. Rogaeva, M. Mullan and P.H. St George-Hyslop, 2000. Molecular genetics of Alzheimer's disease: The role of beta-amyloid and presenilins. Curr. Opin. Neurol., 13: 377-384.
PubMedDirect Link - Rogaeva, E.A., K.C. Fafel, Y.Q. Song, H. Medeiros and C. Sato et al., 2001. Screening for PS1 mutations in a referral-based series of AD cases 21 novel mutations. Neurology, 57: 621-625.
Direct Link - Axelman, K., H. Basun and L. Landfelt, 1998. Wide range of disease onset in a family with Alzheimer disease and a His163Tyr mutation in the presenilin-1 gene. Arch. Neurol., 55: 698-702.
Direct Link