
Ali Vaziri Gohar
Department of Biochemistry, Afzalipour School of Medicine, Kerman University of Medical Sciences, Kerman, Iran
Journal of Biological Sciences
Year: 2010 | Volume: 10 | Issue: 5 | Page No.: 373-385







ABSTRACT
The best characterized of epigenetic is the modifications that occur on DNA and histones that can specify regulate transcriptional activity. Since discovery of cancer epigenetic in 1983, there is an explosion of interest in the epigenetic of cancer study. The epigenetic of cancer has been associated with all stages of tumor formation and progression. DNA methylation and histone acetylation are important epigenetic mechanisms of gene regulation and play essential roles in tumor initiation and progression. Also, epigenetic alterations are potentially reversible, but epigenetic events can facilitate genetic damage by the increased mutagenicity of 5-methylcytosine by DNA methylation. The advantages of epigenetic changes can be used as powerful marker to detect cancer cells or cancer-derived and have made the way of innovative diagnostic and therapeutic strategies, although only a few genes have given promising results as potential tumor biomarkers.
PDF Abstract XML References Citation
Received: March 14, 2010;
Accepted: May 21, 2010;
Published: July 14, 2010
How to cite this article
Ali Vaziri Gohar, 2010. Clinical Applications of Epigenetic Markers in Diagnosis and Treatment of Cancer. Journal of Biological Sciences, 10: 373-385.
DOI: 10.3923/jbs.2010.373.385
URL: https://scialert.net/abstract/?doi=jbs.2010.373.385
DOI: 10.3923/jbs.2010.373.385
URL: https://scialert.net/abstract/?doi=jbs.2010.373.385
INTRODUCTION
Epigenetic changes, unlike genetic alterations defined as heritable changes in gene expression that are potentially reversible and have essential roles during embryonic development, however appear to contribute to the malignant transformation and progression of cancer (Wolffe and Matzke, 1999; Li, 2002). There are three main types of epigenetic information in the genome which are having important role in the regulation of gene expression as follow. Firstly, DNA methylation that is a covalent modification of DNA in which a methyl group is transferred from S-adenosyl-methionine to the C-5 position of cytosine by a family of cytosine methyl-transferases (DNAMTs). DNA methylation occurs almost exclusively at CpG nucleotides. Secondly, Histone modifications which are including acetylation, methylation and phosphorylation which are stably maintained during cell division. Finally, Genomic imprinting which is parent-of-origin-specific allele silencing or relative silencing of one parental allele compared with the other parental allele (Vaziri Gohar and Mohammadi, 2010).
The goal of this study is the background, promise, status of the applications of epigenetic alterations for the early detection, prevention, diagnostics, therapeutics and risk prediction of cancer.
DNA methylation: DNA methylation in mammalian genomes is a defense mechanism by which repetitive DNA which accounts for at least 50% of genome’s content is transcriptionally silenced to prevent it from propagating (Urnov, 2002).
Methyl-cytosine residues are often found in short stretches of CpG-rich regions (CpG islands) that are 0.5-2 kb long and found in the 5′-region of approximately 60% of genes (Gardiner-Garden and Frommer, 1987). When a CpG site is methylated, cytosines on both DNA strands are methylated. At DNA replication, the methylated status is transmitted to daughter DNAs by maintenance DNA methyl-transferase (DNMT), which is present at a replication fork and recognizes hemi-methylated CpG sites. Although a newly synthesized DNA strand does not have methyl groups, maintenance DNA methyl-transferase at the replication fork transfers a methyl group to the newly synthesized strand. Therefore, the methylated or unmethylated status of CpG sites is faithfully copied into daughter DNA in somatic cells (Jones and Baylin, 2002; Herman and Baylin, 2003).
Most CpG islands are unmethylated, with the exception of certain imprinted genes and genes on the inactive X chromosomes of females (Bird, 1986). DNA methylation aberrations can occur as either hypo- or hypermethylation. Both forms can lead to chromosomal instability and transcriptional gene silencing (Baylin et al., 1991).
DNMT1 is referred to as the maintenance methylase due to its preference for hemimethylated CpG sites in DNA (Pradhan et al., 1999). DNMT3a and DNMT3b are considered to be de novo methylases because they can methylate unmethylated DNA (Okano et al., 1999; Pradhan et al., 1999). However, all three DNMTs have been shown to act cooperatively and the functional differences between the methylases may to a large extent be due to the genomic regions that they act upon (Liang et al., 2002; El-Osta, 2003). Over-expression of the DNA methyltransferases 1 and 3A was found in the bone marrow of patients with myelodysplastic syndrome (MDS). Upregulation of DNMTs has also been shown in prostate cancer cell lines and tissues (Patra et al., 2002; Langer et al., 2005).
For detection of cancer cells in body fluids, a high-sensitivity method is necessary. One way is mutation detection in cells. Indeed, because the exact location of a mutation within a gene is usually unknown, many primer sets are necessary for complete analysis. In contrast, aberrant methylation of DNA molecule of cancer cells, even in very few in number, can be sensitively detected by using Methylation-Specific PCR method (MSP), only with one set of PCR primer can be performed on chemically stable DNA, not on RNA (Herman et al., 1996; Laird, 2003).
DNA hypermethylation: Methylation of unmethylated DNA, also known as hypermethylation, can repress the gene expression. The gene repression is caused by changes in chromatin structures due to binding of specific proteins to methylated DNA. This binding leads to decreased affinity for binding of some transcriptional factors to methylated CpG sites. Inappropriate silencing of genes can contribute to cancer initiation, progression, pathologic grade, invasion and metastasis (Ushijima et al., 2003; Laird et al., 2004). Nearly, all types of cancers have transcriptional inactivation of tumor suppressor genes due to DNA hypermethylation (Costello et al., 2000; Herman and Baylin, 2003; Ransohoff, 2003; Jones and Baylin, 2007).
DNA hypomethylation: Demethylation of normally methylated DNA, also known as hypomethylation, can disrupt such a defense mechanism, leading to structural and functional alterations of the genome. There are two types of hypomethylation: global and gene-specific hypomethylation, which refers to an overall decrease of 5-methyl-cytosine content in the genome or a gene, respectively (Dunn, 2003). Both global- and gene-specific hypomethylation have hazardous effects and may contribute in human cancer, like DNA hypermethylation. Global DNA hypomethylation has also been found in the premalignant or early stages of some neoplasms. Also, net decreases in the content of methyl-cytosines, often exceed the localized increases in DNA methylation. However, it is unclear whether this epigenetic alteration is a cause or consequence of tumorigenesis and also, whether hypomethylation induced by disrupting DNMT1 does inhibit or promote tumor growth (Feinberg et al., 1988; Cravo et al., 1996; Baylin et al., 2000; Robertson, 2001).
In a murine model of intestinal neoplasia, mice carrying a germ-line mutation in the APC gene (APCMin/_) crossed with mice heterozygous for the DNMT1 mutation had substantially fewer tumors than Min mice with wild-type DNMT1 (Laird et al., 1995; Cormier and Dove, 2000). In contrast, genomic hypomethylation has been associated with the induction of T-cell lymphomas in mice carrying a hypomorphic DNMT1 allele, which reduces DNMT1 expression to 10% of wild-type levels and results in substantial genome-wide hypomethylation in all tissues. So, hypomethylation-induced cancer might be related to differences in model systems or tissue specificity (Lengauer, 2003).
It is possible that demethylation could lead to the activation proto-oncogenes (Ehrlich, 2002; Fruhwald and Plass, 2002; Nishigaki et al., 2005). Also, genome-wide demethylation may promote genomic instability possibly by activating global demethylation of repetitive sequences such as satellite DNAs can lead to increased chromosomal rearrangements (Dunn, 2003).
Compared with adjacent normal tissues, different types of cancer tissues contain hypomethylated c-jun and c-MYC proto-oncogenes in liver cancer (Tsujiuchi et al., 1999; Tao et al., 2000), pS2 gene in breast cancer (Fruhwald and Plass, 2002) and PLAU gene in the prostate cancer (Van Veldhuizen et al., 1996).
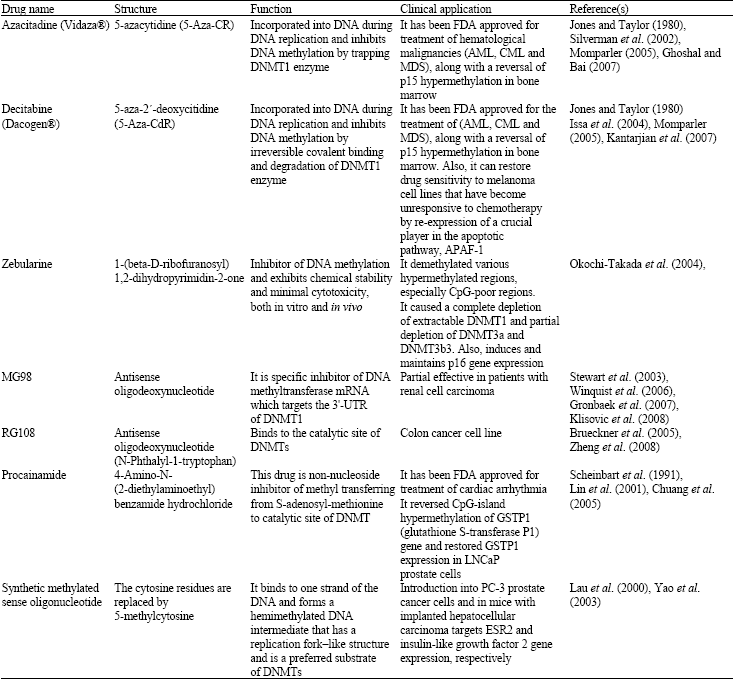
Demethylating agents: Considering that some aberrant DNA methylation is present in early stages of carcinogenesis, there is a possibility that such demethylating agents may protect against some cancers (Laird et al., 1995). Demethylating agents are including DNMT1 inhibitors group (Azacitadine, Decitabine, Zebularine and MG98), procainamide, procaine and EGCG (epigallocatechin-3-gallate) (Fang et al., 2003; Villar-Garea et al., 2003). Inhibitors of DNMTs have been widely used in cell culture systems to reverse abnormal DNA hypermethylation and restore silenced gene expression. However, only limited success has been achieved in clinical trials with these drugs (Thibault et al., 1998; Goffin and Eisenhauer, 2002). Also, nucleosides analog inhibitors of DNMTs may promote genomic instability and increase the risk of cancer in other tissues, because have many potential side effects such as myelotoxicity, mutagenesis and tumorigenesis (Jones and Taylor, 1980; Jackson-Grusby et al., 1997; Gaudet et al., 2003). So, there is an attractive alternative for possible clinical use of non-nucleoside analog DNMT inhibitors.
The use of these drugs raises questions regarding their potential to affect non-cancerous cells epigenetically. However, normal cells divide at a slower rate than malignant cells and incorporate less of these drugs into their DNA resulting in less of an effect on DNA methylation. As shown in Table 1, methylation pattern of multiple genes have higher role in diagnostic and prognostic possibilities than that of a single gene. However, long-term negative effects of DNA methylation inhibitors in patients have not been found to date (Yang et al., 2003).
Azacitadine and decitabine are labil and have acute hematological toxicities. A next generation DNA methylation inhibitor, such as zebularine, might possibly overcome these problems (Marquez et al., 2005; Yoo et al., 2008). Also, the non-nucleoside analogue inhibitors are not as potent as the nucleoside analogues and therefore this issue needs for improvement (Chuang et al., 2005).
DNA methylation as a marker: It has been reported that the pattern of aberrant methylation of individual- or multiple genes can be associated with clinically useful information, such as cancer risk assessment, cancer prognoses, early detection and responses to therapeutics. Therefore, these features make DNA hypermethylation an excellent tumor biomarker candidate (Herman and Baylin, 2003).
In Table 2, aberrant DNA methylation can be applied to detect cancer cells or cancer-derived DNA cancer and diagnostics in several ways. Firstly, if aberrant methylation of some CGIs is specifically present in cancer cells, it can be used to detect cancer cells in biopsy samples or cancer-derived free DNA in plasma. On the other hand, incidences of aberrant methylation of specific CGIs are higher than those of mutations.
| Table 1: | DNA methylation-related drugs |
 | |
| Table 2: | Clinical association of DNA methylation in diagnostic and prognostic of cancer |
 | |
Secondly, if aberrant methylation of some CGIs is associated with a disease phenotype, such as prognosis, responses to chemotherapies or occurrence of adverse effects, it can be used as a marker to predict the phenotype. Finally, if aberrant methylation of some CGIs in non-cancerous tissue is associated with a risk for cancer development, it can be used as a cancer risk marker (Kaneda et al., 2002; Miyamoto et al., 2003; Hagihara et al., 2004).
Histone modification: The N-terminal tails of histones, positioned peripheral to the nucleosome core, are subject to various covalent modifications, such as acetylation, methylation, phosphorylation and ubiquitination by specific chromatin modifying enzymes (Zhang and Reinberg, 2001).
The pattern of these modifications has been referred to as the histone code, and it acts as a second layer of epigenetic regulation of gene expression affecting chromatin structure and remodeling (Jenuwein and Allis, 2001). Histone modification is closely associated with DNA methylation status and is important for gene regulation (Jaenisch and Bird, 2003).
Histone methylation: Histone methylation of histone H3-k4 (Lys4) is associated with active gene transcription. But methylation of histone H3-k9 (Lys9) and H3K27 are normally present at transcriptionally inactive or heterochromatic regions is associated with gene repression (Cao et al., 2002; Nguyen et al., 2002). Lysine residues (lys) can accept up to three methyl groups, which are added by various histone methyltransferases (HMTs) (Fischle et al., 2003; Santos-Rosa and Caldas, 2005).
The methyltransferase MLL, which methylates H3K4, is involved in translocations that lead to the inappropriate expression of various homeotic (Hox) genes, which contributes to leukemic progression (Krivtsov and Armstrong, 2007).
The Polycomb group (PcG) complexes are chromatin modifiers that are crucial to development and have been implicated in the development of cancer (Tonini et al., 2008). These negative regulators of gene expression are very important in sustaining the repressive state of their target genes through the cell cycle (Kingston et al., 1996). Two of the PcG repressive complexes (PRC1 and PRC2) have both been shown to be involved in various cancers. Enhancer of zeste homologue 2 (EZH2), a component of PRC2 with H3K27 methyltransferase activity, is upregulated in mantle cell lymphoma, breast and prostate cancer (Visser et al., 2001; Kleer et al., 2003). RING1, a component of PRC1 that aids in the ubiquitylation of histone H2A lysine 119, is upregulated in prostate cancer (Van Leenders et al., 2007).
Histone acetylation: Histone acetylation is catalyzed by histone acetyltransferases (HATs) and associated with active gene transcription. The basic charges of the histone tails become neutralized upon acetylation. This causes increased accessibility for further modifications or access to the DNA for binding factors and transcriptional machinery (Hebbes et al., 1992; Turner, 1993; Kouzarides, 2007).
Histone deacetylation, mediated by three classes of HDACs which remove the acetyl group from lysine residues and associated with active gene transcription (Gray and Ekstrom, 2001; Yoshida et al., 2001; Marks et al., 2004). Inappropriate deacetylation can also contribute to cancer progression. HDACs are upregulated in various types of cancer, such as gastric, prostate, oral squamous cell and lung. Over-expression of HDACs can also lead to the transcriptional inactivation of tumor suppressors, such as p53 (Halkidou et al., 2004; Bartling et al., 2005; Song et al., 2005; Sakuma et al., 2006).
HDAC inhibitors are divided into 4 groups based on their structures including hydroxamic acids, cyclic peptides, short chain fatty acids and benzamides (Zheng et al., 2008). HDAC inhibitors have pleiotropic effects including inhibition of angiogenesis, induction of apoptosis and cell cycle arrest (Stearns et al., 2007). The hydroxamic acid group of HDAC inhibitors has been successful in treating both hematologic malignancies and solid tumors. The X-ray crystallography has shown that the catalytic site of HDACs contains a zinc atom. The hydroxamic acid can fit into the catalytic site of HDACs and bind to the zinc atom of this site thereby inhibiting the HDAC (Marks et al., 2000). The shortcoming of these HDAC inhibitors is that a high concentration of drug is required for efficacy resulting in limited use in the clinic (Johnstone, 2002). The list of the HDAC inhibitory agents was shown in Table 3.
| Table 3: | The anti-cancer drugs which exhibit HDAC inhibitory activity |
 | |
Neither of these drugs is as potent as the other classes of HDAC inhibitors and seems to have the greatest effect when used in a combinatorial treatment (Kouraklis and Theocharis, 2006). For example, sodium butyrate and TSA synergize with 1,25-(OH)2-vitamin D3 to inhibit the growth of LNCaP, PC-3 and DU-145 prostate cancer cells by inducing apoptosis (Fruhwald and Plass, 2002).
In prostate cancer, the expression of several genes may be potentially regulated by histone acetylation. Treatment of prostate cancer cells with HDAC inhibitors increased expression of specific genes such as insulin-like growth factor-binding protein 3 (Tsubaki et al., 2002) and carboxypeptidase A3 (Huang et al., 1999) and thus inferred a role for histone acetylation in gene regulation.
One such gene, coxsackie and adenovirus receptor (CAR), is the primary receptor for group C adenoviruses and is important for adenovirus attachment to the cell membrane. In urogenital cancer cells, including the prostate cancer cell line PC-3, activation of the CAR gene is modulated by histone acetylation and can be induced by depsipeptide, an HDAC inhibitor (Pong et al., 2003). Exposing cancer cells to low concentrations of depsipeptide has the functional consequence of preferentially increasing the efficiency of adenoviral transgene expression (Goldsmith et al., 2003).
Another gene regulated by histone modification is the vitamin D receptor. 1,25-(OH)2-vitamin D3 acts to antiproliferative effects in a variety of tumor cells, including those of the prostate (Moffatt et al., 1999; Zhao et al., 2000; Ikeda et al., 2003; Yang and Burnstein, 2003).
Prostate cancer cells that are insensitive to 1,25-(OH)2-vitamin D3 have increased levels of nuclear receptor corepressor SMRT (silencing mediator of retinoid and thyroid), which could result in increased deacetylase activity and decreased transcriptional activity of the vitamin D receptor. In addition, combined treatment of prostate cancer cell lines with the HDAC inhibitor trichostatin A (TSA) and 1,25-(OH)2-vitamin D3 synergistically inhibits cell proliferation. This finding may be useful in the clinical setting, in which use of 1,25-(OH)2-vitamin D3 and its analogs in combination with HDAC inhibitors could activate the vitamin D receptor while minimizing unwanted side effects associated with 1,25-(OH)2- vitamin D3, such as hypocalcemia (Banwell et al., 2003).
Loss of imprinting (LOI): Also, aberrant methylation of imprinted genes can disturb imprinting. LOI of insulin like growth factor 2 (IGF2) is causally involved in Wilms tumors and colorectal cancers through its overexpression (Feinberg and Tycko, 2004). Loss of IGF2 imprinting in colonic mucosae is associated with an elevated risk of colorectal cancers (Cui et al., 1998, 2003; Woodson et al., 2004) and LOI in peripheral lymphocyte was also associated with an increased risk (Cui et al., 2003). A study using a mouse model for loss of IGF2 imprinting showed that LOI caused less differentiation of normal intestinal epithelium (Sakatani et al., 2005).
DISCUSSION
The DNA methylation and histone modifications are as important interconnected epigenetic regulatory mechanisms that they can influence the gene regulation (Fuks et al., 2003). DNA methylation is involved in gene silencing through binding of methylated DNA binding proteins such as MeCP2 to gene promoter. This interaction then recruits HDAC to methylated promoters. Therefore, DNA methylation event happens first followed by histone deacetylation and then histone methylation (Antequera and Bird, 1999; Stirzaker et al., 2004).
Several studies have shown that the combination of HDAC and DNMT inhibitors can work synergistically to induce the re-expression of such genes like tumor suppressor genes and genes that involved in apoptosis, differentiation, cell growth arrest could enhance the antitumor effects, in vitro (Cameron et al., 1999; Weiser et al., 2001; Ghoshal et al., 2002). For example, phenylbutyrate and 5-Aza-CdR have synergistic effects on reducing lung tumor formation in mice more than 5-Aza-CdR alone (Belinsky et al., 2003). So, DNA methylation inhibitors and HDAC inhibitors are now used together in the clinic after garnering encouraging results in vitro and it is hypothesized that in a phase II trial, using a longer exposure could further increase the response rate (Gore et al., 2006). However, non-specific demethylation has the risk of inducing demethylation of normally methylated sequences, such as retrotransposons and thus retrotranspositions.
Pretreatment with an HDAC inhibitor can greatly increase cytotoxicity in various cell lines when followed by subsequent treatment of a chemotherapeutic drug (kim et al., 2003). Likewise, cisplatin resistant cells from head and neck cancer cell lines can be reprogrammed to become responsive after treatment with Phenyl butyrate (Burkitt and Ljungman, 2008). The increased sensitivity to other drugs after use of an epigenetic drug is encouraging since drug resistance does present a challenge in effective cancer treatment.
CONCLUSIONS
As the field of cancer epigenetic advances, a better understanding of the DNA methylation and post-translational histone modifications which play central roles in gene regulation is under developing. The future directions for the development of epigenetic drugs and epigenetic markers will depend on the elucidation of their mechanisms and the downstream effects of treatment.
The use of FDA approved epigenetic drugs has gained momentum and has proven useful in some tumors. The combinatorial use of DNA methylation inhibitors, HDAC inhibitors and non-epigenetic chemotherapeutic drugs in an effort increase response rates and maximize the efficacy of these drugs in the clinic and may have synergistic effects in re-establishing the expression of tumor suppressor genes. However, much work remains in designing drugs that will be more stable, less toxic and more specific in their enzyme inhibition. Future epigenetic drugs can also be designed to target histone methyltransferases, histone demethylases or other chromatin modifiers not yet discovered.
ACKNOWLEDGMENT
I am grateful to all the research scientists, especially to Professors. Andrew Feinberg and Stephen Baylin from School of Medicine at Johns Hopkins University for several years studies on cancer epigenetic.
REFERENCES
- Ahrendt, S.A., J.T. Chow, L.H. Xu, S.C. Yang and C.F. Eisenberger et al., 1999. Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J. Natl. Cancer Inst., 91: 332-339.
PubMed - Antequera, F. and A. Bird, 1999. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr. Biol., 9: R661-667.
CrossRef - Banwell, C.M., R. Singh, P.M. Stewart, M.R. Uskokovic and M.J. Campbell, 2003. Antiproliferative signalling by 1,25(OH)2D3 in prostate and breast cancer is suppressed by a mechanism involving histone deacetylation. Recent Results Cancer Res., 164: 83-98.
PubMed - Bartling, B., H.S. Hofmann, T. Boettger, G. Hansen, S. Burdach, R.E. Silber and A. Simm, 2005. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer, 49: 145-154.
PubMed - Battagli, C., R.G. Uzzo, E. Dulaimi, I.I. de Caceres and R. Krassenstein et al., 2003. Promoter hypermethylation of tumor suppressor genes in urine from kidney cancer patients. Cancer Res., 63: 8695-8699.
Direct Link - Baylin, S.B., M. Makos, J.J. Wu, R.W. Yen and A. De Bustros et al., 1991. Abnormal patterns of DNA methylation in human neoplasia: potential consequences for tumor progression. Cancer Cells, 3: 383-390.
PubMed - Baylin, S.B., S.A. Belinsky and J.G. Herman, 2000. Aberrant methylation of gene promoters in cancer-concepts, misconcepts, and promise. J. Natl. Cancer Inst., 92: 1460-1461.
CrossRefDirect Link - Belinsky, S.A., D.M. Klinge, C.A. Stidley, J.P. Issa, J.G. Herman, T.H. March and S.B. Baylin, 2003. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res., 63: 7089-7093.
Direct Link - Belinsky, S.A., K.J. Nikula, W.A. Palmisano, R. Michels and G. Saccomanno et al., 1998. Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc. Natl. Acad. Sci., 95: 11891-11896.
Direct Link - Belinsky, S.A., W.A. Palmisano, F.D. Gilliland, L.A. Crooks and K.K. Divine et al., 2001. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res., 62: 2370-2377.
PubMed - Blaheta, R.A., M. Michaelis, P.H. Driever and J. Cinatl, 2005. Evolving anticancer drug valproic acid: Insights into the mechanism and clinical studies. Med. Res. Rev., 25: 383-397.
PubMed - Brueckner, B., R.G. Boy, P. Siedlecki, T. Musch and H.C. Kliem et al., 2005. Epigenetic reactivation of tumor suppressor genes by a novel small-molecule inhibitor of human DNA methyltransferases. Cancer Res., 65: 6305-6311.
Direct Link - Burkitt, K. and M. Ljungman, 2008. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol. Cancer, 7: 24-24.
Direct Link - Butler, L.M., Y. Webb, D. B. Agus, B. Higgins and T.R. Tolentino et al., 2001. Inhibition of transformed cell growth and induction of cellular differentiation by pyroxamide, an inhibitor of histone deacetylase. Clin. Cancer Res., 7: 962-970.
Direct Link - Cairn, P., M. Esteller, J.G. Herman, M. Schoenberg and C. Jeronimo et al., 2001. Molecular detection of prostate cancer in urine byGSTP1 hypermethylation. Clin. Cancer Res., 7: 2727-2730.
PubMed - Camacho, L.H., J. Olson, W.P. Tong, C.W. Young, D.R. Spriggs and M.G. Malkin, 2007. Phase I dose escalation clinical trial of phenylbutyrate sodium administered twice daily to patients with advanced solid tumors. Invest. New Drugs, 25: 131-138.
CrossRef - Cameron, E.E., K.E. Bachman, S. Myohanen, J.G. Herman and S.B. Baylin, 1999. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet., 21: 103-107.
CrossRefDirect Link - Camphausen, K., T. Scott, M. Sproull and P.J. Tofilon, 2004. Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin. Cancer Res., 10: 6066-6071.
CrossRef - Cao, R., L. Wang, H. Wang, L. Xia and H. Erdjument-Bromage et al., 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science, 298: 1039-1043.
PubMed - Chan, M.W., L.W. Chan, N.L. Tang, J.H. Tong and K.W. Lo et al., 2002. Hypermethylation of multiple genes in tumor tissues and voided urine in urinary bladder cancer patients. Clin. Cancer Res., 8: 464-470.
Direct Link - Chuang, J.C., C.B. Yoo, J.M. Kwan, T.W. Li, G. Liang, A.S. Yang and P.A. Jones, 2005. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2-deoxycytidine. Mol. Cancer Ther., 4: 1515-1520.
CrossRef - Cinatl, J., R. Kotchetkov, R. Blaheta, P.H. Driever, J.U. Vogel and J. Cinatl, 2002. Induction of differentiation and suppression of malignant phenotype of human neuroblastoma BE(2)-C cells by valproic acid: Enhancement by combination with interferon-alpha. Int. J. Oncol., 20: 97-106.
PubMed - Cormier, R.T. and W.F. Dove, 2000. Dnmt1N/_ reduces the net growth rate and multiplicity of intestinal adenomas in C57BL/6-multiple intestinal neoplasia (Min)/_ mice independently of p53 but demonstrates strong synergy with the modifier of Min 1(AKR) resistance allele. Cancer Res., 60: 3965-3970.
PubMed - Costello, J.F., C. Plass and W.K. Cavenee, 2000. Aberrant methylation of genes in low-grade astrocytomas. Brain Tumor Pathol., 17: 49-56.
CrossRef - Cravo, M., R. Pinto, P. Fidalgo, P. Chaves and L. Gloria et al., 1996. Global DNA hypomethylation occurs in the early stages of intestinal type gastric carcinoma. Gut, 39: 434-438.
CrossRef - Cui, H., I.L. Horon, R. Ohlsson, S.R. Hamilton and A.P. Feinberg, 1998. Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability. Nat. Med., 4: 1276-1280.
CrossRef - Cui, H., M. Cruz-Correa, F.M. Giardiello, D.F. Hutcheon and D.R. Kafonek et al., 2003. Loss of IGF2 imprinting: A potential marker of colorectal cancer risk. Science, 299: 1753-1755.
CrossRef - Dulaimi, E., R.G. Uzzo, R.E. Greenberg, T. Al-Saleem and P. Cairns, 2004. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin. Cancer Res., 10: 1887-1893.
CrossRef - Dunn, B.K., 2003. Hypomethylation: One side of a larger picture. Ann. N. Y. Acad. Sci., 983: 28-42.
PubMed - Dyer, E.S., M.T. Paulsen, S.M. Markwart, M. Goh, D.L. Livant and M. Ljungman, 2002. Phenylbutyrate inhibits the invasive properties of prostate and breast cancer cell lines in the sea urchin embryo basement membrane invasion assay. Int. J. Cancer, 101: 496-499.
PubMed - Ehrlich, M., 2002. DNA methylation in cancer: Too much, but also too little. Oncogene, 21: 5400-5413.
Direct Link - El-Osta, A., 2003. DNMT cooperativity the developing links between methylation, chromatin structure and cancer. Bioessays, 25: 1071-1084.
PubMed - Esteller, M., J. Garcia-Foncillas, E. Andion, S.N. Goodman and O.F. Hidalgo et al., 2000. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med., 343: 1350-1354.
Direct Link - Esteller, M., J.M. Silva, G. Dominguez, F. Bonilla and Xavier Matias-Guiu et al., 2000. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer. Inst., 92: 564-569.
CrossRef - Evron, E., W.C. Dooley, C.B. Umbricht, D. Rosenthal and N. Sacchi et al., 2001. Detection of breast cancer cells in ductal lavage fluid by methylationspecific PCR. Lancet, 357: 1335-1336.
PubMed - Fang, M.Z., Y. Wang, N. Ai, Z. Hou and Y. Sun et al., 2003. Tea polyphenol (-) epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res., 63: 7563-7570.
Direct Link - Feinberg, A.P. and B. Tycko, 2004. The history of cancer epigenetics. Nat. Rev. Cancer, 4: 143-153.
PubMed - Feinberg, A.P., C.W. Gehrke, K.C Kuo and M. Ehrlich, 1988. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res., 48: 1159-1161.
PubMed - Fischle, W., Y. Wang and C.D. Allis, 2003. Binary switches and modification cassettes in histone biology and beyond. Nature, 425: 475-479.
CrossRefDirect Link - Fruhwald, M.C. and C. Plass, 2002. Global and gene-specific methylation patterns in cancer: Aspects of tumor biology and clinical potential. Mol. Genet. Metab., 75: 1-16.
PubMed - Fuks, F., P.J. Hurd, D. Wolf, X. Nan, A.P. Bird and T. Kouzarides, 2003. The methyl- CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem., 278: 4035-4040.
CrossRef - Gardiner-Garden, M. and M. Frommer, 1987. CpG islands in vertebrate genomes. J. Mol. Biol., 196: 261-282.
PubMed - Gaudet, F., J.G. Hodgson, A. Eden, L. Jackson-Grusby and J. Dausman et al., 2003. Induction of tumors in mice by genomic hypomethylation. Science, 300: 489-492.
CrossRef - Ghoshal, K., J. Datta, S. Majumder, S. Bai and X. Dong et al., 2002. Inhibitors of histone deacetylase and DNA methyltransferase synergistically activate the methylated metallothionein I promoter by activating the transcription factor MTF-1 and forming an open chromatin structure. Mol. Cell. Biol., 22: 8302-8319.
CrossRefDirect Link - Ghoshal, K. and S. Bai, 2007. DNA methyltransferases as targets for cancer therapy. Drugs Today (Barc), 43: 395-422.
CrossRef - Glaser, K.B., 2007. HDAC inhibitors: Clinical update and mechanism-based potential. Biochem. Pharmacol., 74: 659-671.
PubMed - Goessl, C., H. Krause, M. Muller, R. Heicappell and M. Schrader et al., 2000. Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res., 60: 5941-5945.
Direct Link - Goffin, J. and E. Eisenhauer, 2002. DNA methyltransferase inhibitors-state of the art. Ann. Oncol., 13: 1699-1716.
CrossRef - Goldsmith, M.E., M. Kitazono, P. Fok, T. Aikou, S. Bates and T. Fojo, 2003. The histone deacetylase inhibitor FK228 preferentially enhances adenovirus transgene expression in malignant cells. Clin. Cancer Res., 9: 5394-5401.
Direct Link - Gore, S.D., L.J. Weng, W.D. Figg, S. Zhai and R.C. Donehower et al., 2002. Impact of prolonged infusions of the putative differentiating agent sodium phenylbutyrate on myelodysplastic syndromes and acute myeloid leukemia. Clin. Cancer Res., 8: 963-970.
Direct Link - Gore, S.D., S. Baylin, E. Sugar, H. Carraway and C.B. Miller et al., 2006. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res., 66: 6361-6369.
CrossRef - Gray, S.G. and T.J. Ekstrom, 2001. The human histone deacetylase family. Exp. Cell Res., 262: 75-83.
PubMed - Gronbaek, K., C. Hother and P.A. Jones, 2007. Epigenetic changes in cancer. Apmis, 115: 1039-1059.
PubMed - Hagihara, A., K. Miyamoto, J. Furuta, N. Hiraoka and K. Wakazono et al., 2004. Identification of 27 50 CpG islands aberrantly methylated and 13 genes silenced in human pancreatic cancers. Oncogene, 23: 8705-8710.
CrossRef - Halkidou, K., L. Gaughan, S. Cook, H.Y. Leung, D.E. Neal and C.N. Robson, 2004. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate, 59: 177-189.
PubMed - Hebbes, T.R., A.W. Thorne, A.L. Clayton and C. Crane-Robinson, 1992. Histone acetylation and globin gene switching. Nucleic Acids Res., 20: 1017-1022.
Direct Link - Herman, J.G. and S.B. Baylin, 2003. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med., 349: 2042-2054.
PubMed - Herman, J.G., J.R. Graff, S. Myohanen, B.D. Nelkin and S.B. Baylin, 1996. Methylation specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci., 93: 9821-9826.
Direct Link - Hibi, K., M. Taguchi, H. Nakayama, T. Takase and Y. Kasai et al., 2001. Molecular detection of p16 promoter methylation in the serum of patients with esophageal squamous cell carcinoma. Clin. Cancer Res., 7: 3135-3138.
Direct Link - Huang, H., C.P. Reed, J.S. Zhang, V. Shridhar, L. Wang and D.I. Smith, 1999. Carboxypeptidase A3 (CPA3): A novel gene highly induced by histone deacetylase inhibitors during differentiation of prostate epithelial cancer cells. Cancer Res., 59: 2981-2988.
PubMed - Ikeda, N., H. Uemura, H. Ishiguro, M. Hori and M. Hosaka et al., 2003. Combination treatment with 1alpha, 25-dihydroxyvitamin D3 and 9-cisretinoic acid directly inhibits human telomerase reverse transcriptase transcription in prostate cancer cells. Mol. Cancer Ther., 2: 739-746.
PubMed - Issa, J.P., G. Garcia-Manero, F.J. Giles, R. Mannari and D. Thomas et al., 2004. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-20-deoxycytidine (decitabine) in hematopoietic malignancies. Blood, 103: 1635-1640.
PubMed - Jackson-Grusby, L., P.W. Laird, S.N. Magge, B.J. Moeller and R. Jaenisch, 1997. Mutagenicity of 5-aza-2_-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proc. Natl. Acad. Sci., 94: 4681-4685.
PubMed - Jaenisch, R. and A. Bird, 2003. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet., 33: 245-254.
CrossRef - Jenuwein, T. and C.D. Allis, 2001. Translating the histone code. Science, 293: 1074-1080.
CrossRefDirect Link - Johnstone, R.W., 2002. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov., 1: 287-299.
CrossRefPubMedDirect Link - Jones, P.A. and S.B. Baylin, 2002. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet., 3: 415-428.
CrossRefPubMedDirect Link - Jones, P.A. and S.M. Taylor, 1980. Cellular differentiation, cytidine analogs and DNA methylation. Cell, 20: 85-93.
PubMed - Kaneda, A., M. Kaminishi, K. Yanagihara, T. Sugimura and T. Ushijima, 2002. Identification of silencing of nine genes in human gastric cancers. Cancer Res., 62: 6645-6650.
Direct Link - Kantarjian, H.M., S. O'Brien, J. Shan, A. Aribi and G. Garcia-Manero et al., 2007. Update of the decitabine experience in higher risk myelodysplastic syndrome and analysis of prognostic factors associated with outcome. Cancer, 109: 265-273.
Direct Link - Kawakami, K., J. Brabender, R.V. Lord, S. Groshen and B.D. Greenwald et al., 2000. Hypermethylated APC DNA in plasma and prognosis of patients with esophageal adenocarcinoma. J. Natl. Cancer Inst., 92: 1805-1811.
CrossRef - Kersting, M., C. Friedl, A. Kraus, M. Behn, W. Pankow and M. Schuermann, 2000. Differential frequencies of p16(INK4a) promoter hypermethylation, p53 mutation, and K-ras mutation in exfoliative material mark the development of lung cancer in symptomatic chronic smokers. J. Clin. Oncol., 18: 3221-3229.
PubMed - Kim, M.S., M. Blake, J.H. Baek, G. Kohlhagen, Y. Pommier and F. Carrier, 2003. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res., 63: 7291-7300.
PubMedDirect Link - Kingston, R.E., C.A. Bunker and A.N. Imbalzano, 1996. Repression and activation by multiprotein complexes that alter chromatin structure. Genes Dev., 10: 905-920.
PubMed - Kleer, C.G., Q. Cao, S. Varambally, R. Shen and I. Ota et al., 2003. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci., 100: 11606-11611.
CrossRef - Klisovic, R.B., W. Stock, S. Cataland, M.I. Klisovic and S. Liu et al., 2008. A phase I biological study of MG98, an oligodeoxynucleotide antisense to DNA methyltransferase 1, in patients with high-risk myelodysplasia and acute myeloid leukemia. Clin. Cancer Res., 14: 2444-2449.
CrossRef - Kouraklis, G. and S. Theocharis, 2006. Histone deacetylase inhibitors: A novel target of anticancer therapy (review). Oncol. Rep., 15: 489-494.
PubMed - Kouzarides, T., 2007. Chromatin modifications and their function. Cell, 128: 693-705.
CrossRefPubMedDirect Link - Krassenstein, R., E. Sauter, E. Dulaimi, C. Battagli and H. Ehya et al., 2004. Detection of breast cancer in nipple aspirate fluid by CpG island hypermethylation. Clin. Cancer Res., 10: 28-32.
CrossRef - Krivtsov, A.V. and S.A. Armstrong, 2007. MLL translocations histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer, 7: 823-833.
CrossRefDirect Link - Lai, M.T., C.C. Yang, T.Y. Lin, F.J. Tsai and W.C. Chen, 2008. Depsipeptide (FK228) inhibits growth of human prostate cancer cells. Urol. Oncol., 26: 182-189.
CrossRef - Laird, C.D., N.D. Pleasant, A.D. Clark, J.L. Sneeden and K.M. Hassan et al., 2004. Hairpin-bisulfite PCR: Assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc. Natl. Acad. Sci., 101: 204-209.
CrossRef - Laird, P.W., 2003. The power and the promise of DNA methylation markers. Nat. Rev. Cancer, 3: 253-266.
CrossRef - Laird, P., L. Jackson-Grusby, A. Fazeli, S. Dickinson, W. Jung, R. Weinberg and R. Jaenisch, 1995. Suppression of intestinal neoplasia by DNA hypomethylation. Cell, 81: 197-205.
Direct Link - Langer, F., J. Dingemann, H. Kreipe and U. Lehmann, 2005. Up-regulation of DNA methyltransferases DNMT1, 3A, and 3B in myelodysplastic syndrome. Leuk. Res., 29: 325-329.
PubMed - Lau, K.M., M. LaSpina, J. Long and S.M. Ho, 2000. Expression of estrogen receptor (ER)-α and ER-β in normal and malignant prostatic epithelial cells: Regulation by methylation and involvement in growth regulation. Cancer Res., 60: 3175-3182.
PubMed - Li, E., 2002. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet., 3: 662-673.
PubMed - Liang, G., M.F. Chan, Y. Tomigahara, Y.C. Tsai and F.A. Gonzales et al., 2002. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell Biol., 22: 480-491.
PubMed - Lin, X., K. Asgari, M.J. Putzi, W.R. Gage and X. Yu et al., 2001. Reversal of GSTP1 CpG island hypermethylation and reactivation of pi-class glutathione S-transferase (GSTP1) expression in human prostate cancer cells by treatment with procainamide. Cancer Res., 61: 8611-8616.
Direct Link - Marks, P.A., V.M. Richon and R.A. Rifkind, 2000. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst., 92: 1210-1216.
PubMed - Marks, P.A., V.M. Richon, T. Miller, W.K. Kelly, 2004. Histone deacetylase inhibitors. Adv. Cancer Res., 91: 137-168.
CrossRef - Marquez, V.E., J.J. Barchi, J.A. Kelley, K.V. Rao and R. Agbaria et al., 2005. Zebularine: A unique molecule for an epigenetically based strategy in cancer chemotherapy. The magic of its chemistry and biology. Nucleosides Nucleotides Nucleic Acids, 24: 305-318.
PubMed - Miyamoto, K., K. Asada, T. Fukutomi, E. Okochi and Y. Yagi et al., 2003. Methylation-associated silencing of heparan sulfate D-glucosaminyl 3-Osulfotransferase-2 (3-OST-2) in human breast, colon, lung and pancreatic cancers. Oncogene, 22: 274-280.
PubMed - Moffatt, K.A., W.U. Johannes and G.J. Miller, 1999. 1Alpha, 25dihydroxyvitamin D3 and platinum drugs act synergistically to inhibit the growth of prostate cancer cell lines. Clin. Cancer Res., 5: 695-703.
PubMed - Momparler, R.L., 2005. Epigenetic therapy of cancer with 5-aza-2-deoxycytidine (decitabine). Semin. Oncol., 32: 443-451.
PubMed - Muller, H.M., M. Oberwalder, H. Fiegl, M. Morandell and G. Goebel et al., 2004. Methylation changes in faecal DNA: A marker for colorectal cancer screening. Lancet, 363: 1283-1285.
PubMed - Nguyen, C.T., D.J. Weisenberger, M. Velicescu, F.A. Gonzales and J.C. Lin et al., 2002. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2-deoxycytidine. Cancer Res., 62: 6456-6461.
PubMed - Nishigaki, M., K. Aoyagi, I. Danjoh, M. Fukaya and K. Yanagihara et al., 2005. Discovery of aberrant expression of R-RAS by cancer-linked DNA hypomethylation in gastric cancer using microarrays. Cancer Res., 65: 2115-2124.
Direct Link - Okano, M., D.W. Bell, D.A. Haber and E. Li, 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99: 247-257.
CrossRefPubMedDirect Link - Okochi-Takada, E., S. Ichimura, A. Kaneda, T. Sugimura and T. Ushijima, 2004. Establishment of a detection system for demethylating agents using an endogenous promoter CpG island. Mutat. Res., 568: 187-194.
PubMed - Olsen, E.A., Y.H. Kim, T.M. Kuzel, T.R. Pacheco and F.M. Foss et al., 2007. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol., 25: 3109-3115.
PubMed - Palmisano, W.A., K.K. Divine, G. Saccomanno, F.D. Gilliland and S.B. Baylin et al., 2000. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res., 60: 5954-5958.
Direct Link - Pan, L., A.F. Matloob, J. Du, H. Pan and Z. Donget al., 2010. Vitamin D stimulates apoptosis in gastric cancer cells in synergy with trichostatin A /sodium butyrate-induced and 5-aza-2'-deoxycytidine-induced PTEN upregulation. FEBS J., 277: 989-999.
PubMed - Patra, S.K., A. Patra, H. Zhao and R. Dahiya, 2002. DNA methyltransferase and demethylase in human prostate cancer. Mol. Carcinog., 33: 163-171.
PubMed - Pauer, L.R., J. Olivares, C. Cunningham, A. Williams and W. Grove et al., 2004. Phase I study of oral CI-994 in combination with carboplatin and paclitaxel in the treatment of patients with advanced solid tumors. Cancer Invest., 22: 886-896.
PubMed - Pong, R.C., Y.J. Lai, H. Chen, T. Okegawa and E. Frenkel et al., 2003. Epigenetic regulation of coxsackie and adenovirus receptor (CAR) gene promoter in urogenital cancer cells. Cancer Res., 63: 8680-8686.
PubMed - Pradhan, S., A. Bacolla, R.D. Wells and R.J. Roberts, 1999. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem., 274: 33002-33010.
CrossRef - Prakash, S., B.J. Foster, M. Meyer, A. Wozniak and L.K. Heilbrun et al., 2001. Chronic oral administration of CI-994: A phase 1 study. Invest. New. Drugs, 19: 1-11.
CrossRef - Qian, D.Z., Y.F. Wei, X. Wang, Y. Kato, L. Cheng and R. Pili, 2007. Antitumor activity of the histone deacetylase inhibitor MS-275 in prostate cancer models. Prostate, 67: 1182-1193.
PubMed - Ransohoff, D.F., 2003. Cancer: Developing molecular biomarkers for cancer. Science, 299: 1679-1680.
CrossRef - Ricciardiello, L., A. Goel, V. Mantovani, T. Fiorini and S. Fossi et al., 2003. Frequent loss of hMLH1 by promoter hypermethylation leads to microsatellite instability in adenomatous polyps of patients with a single first-degree member affected by colon cancer. Cancer Res., 63: 787-792.
Direct Link - Robertson, K.D., 2001. DNA methylation, methyltransferases and cancer. Oncogene, 20: 3139-3155.
PubMed - Rosas, S.L., W. Koch, M.G. da Costa Carvalho, L. Wu and J. Califano et al., 2001. Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res., 61: 939-942.
Direct Link - Sakatani, T., A. Kaneda, C.A. Iacobuzio-Donahue, M.G. Carter and S. de Boom Witzel, et al., 2005. Loss of imprinting of Igf2 alters intestinal maturation and tumorigenesis in mice. Science, 307: 1976-1978.
PubMed - Sakuma, T., K. Uzawa, T. Onda, M. Shiiba, H. Yokoe, T. Shibahara and H. Tanzawa, 2006. Aberrant expression of histone deacetylase 6 in oral squamous cell carcinoma. Int. J. Oncol., 29: 117-124.
PubMed - Santos-Rosa, H. and C. Caldas, 2005. Chromatin modifier enzymes, the histone code and cancer. Eur. J. Cancer, 41: 2381-2402.
PubMed - Sasakawa, Y., Y. Naoe, T. Noto, T. Inoue and T. Sasakawa et al., 2003. Antitumor efficacy of FK228, a novel histone deacetylase inhibitor, depends on the effect on expression of angiogenesis factors. Biochem. Pharmacol., 66: 897-906.
PubMed - Scheinbart, L.S., M.A. Johnson, L.A. Gross, S.R. Edelstein and B.C. Richardson, 1991. Procainamide inhibits DNA methyltransferase in a human T cell line. J. Rheumatol., 18: 530-534.
PubMed - Silverman, L.R., E.P. Demakos, B.L. Peterson, A.B. Kornblith and J.C. Holland et al., 2002. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: A study of the cancer and leukemia group B. J. Clin. Oncol., 20: 2429-2440.
Direct Link - Song, J., J.H. Noh, J.H. Lee, J.W. Eun and Y.M. Ahn et al., 2005. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS, 113: 264-268.
CrossRef - Soria, J.C., M. Rodriguez, D.D. Liu and J.J. Lee et al., 2002. Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res., 62: 351-355.
Direct Link - Stearns, V., Q. Zhou and N.E. Davidson, 2007. Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest., 25: 659-665.
PubMed - Steele, N.L., J.A. Plumb, L. Vidal, J. Tjornelund and P. Knoblauch et al., 2008. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin. Cancer Res., 14: 804-810.
CrossRef - Stewart, D.J., R.C. Donehower, E.A. Eisenhauer, N. Wainman and A.K. Shah et al., 2003. A phase I pharmacokinetic and pharmacodynamic study of the DNA methyltransferase 1 inhibitor MG98 administered twice weekly. Ann. Oncol., 14: 766-774.
PubMed - Stirzaker, C., J.Z. Song, B. Davidson and S.J. Clark, 2004. Transcriptional gene silencing promotes DNA hypermethylation through a sequential change in chromatin modifications in cancer cells. Cancer Res., 64: 3871-3877.
CrossRef - Suenaga, M., H. Soda, M. Oka, A. Yamaguchi and K. Nakatomi et al., 2002. Histone deacetylase inhibitors suppress telomerase reverse transcriptase mRNA expression in prostate cancer cells. Int. J. Cancer, 97: 621-625.
PubMed - Tao, L., S. Yang, M. Xie, P.M. Kramer and M.A. Pereira, 2000. Hypomethylation and overexpression of c-jun and c-myc protooncogenes and increased DNA methyltransferase activity in dichloroacetic and trichloroacetic acidpromoted mouse liver tumors. Cancer Lett., 158: 185-193.
PubMed - Thelen, P., S. Schweyer, B. Hemmerlein, W. Wuttke, F. Seseke and R.H. Ringert, 2004. Expressional changes after histone deacetylase inhibition by valproic acid in LNCaP human prostate cancer cells. Int. J. Oncol., 24: 25-31.
PubMed - Thibault, A., W.D. Figg, R.C. Bergan, R.M. Lush and C.E. Myers et al., 1998. A phase II study of 5-aza-2deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer. Tumori, 84: 87-89.
PubMed - Tonini, T., G. D'Andrilli, A. Fucito, L. Gaspa and L. Bagella, 2008. Importance of Ezh2 polycomb protein in tumorigenesis process interfering with the pathway of growth suppressive key elements. J. Cell Physiol., 214: 295-300.
CrossRef - Tsubaki, J., V. Hwa, S.M. Twigg and R.G. Rosenfeld, 2002. Differential activation of the IGF binding protein-3 promoter by butyrate in prostate cancer cells. Endocrinology, 143: 1778-1788.
Direct Link - Tsujiuchi, T., M. Tsutsumi, Y. Sasaki, M. Takahama and Y. Konishi, 1999. Hypomethylation of CpG sites and c-myc gene overexpression in hepatocellular carcinomas, but not hyperplastic nodules, induced by a choline-deficient L-amino acid-defined diet in rats. Jpn. J. Cancer. Res., 90: 909-913.
PubMed - Urnov, F.D., 2002. Methylation and the genome: The power of a small amendment. J. Nutr., 132: 2450S-2456S.
PubMed - Ushijima, T., N. Watanabe, E. Okochi, A. Kaneda, T. Sugimura and K. Miyamoto, 2003. Fidelity of the methylation pattern and its variation in the genome. Genome Res., 13: 868-874.
Direct Link - Van Leenders, G.J., D. Dukers, D. Hessels, S.W. Van den Kieboom and C.A. Hulsbergen et al., 2007. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur. Urol., 52: 455-463.
Direct Link - Van Veldhuizen, P.J., R. Sadasivan, R. Cherian and A. Wyatt, 1996. Urokinase-type plasminogen activator expression in human prostate carcinomas. Am. J. Med. Sci., 312: 8-11.
PubMed - Gohar, A.V. and A. Mohammadi, 2010. Epigenetic effects of carcinogens. J. Biol. Sci., 10: 200-208.
CrossRefDirect Link - Villar-Garea, A., M.F. Fraga, J. Espada and M. Esteller, 2003. Procaine is a DNAdemethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res., 63: 4984-4989.
Direct Link - Visser, H.P., M.J. Gunster, H.C. Kluin-Nelemans, E.M. Manders and F.M. Raaphorst et al., 2001. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br. J. Haematol., 112: 950-958.
PubMed - Weiser, T.S., Z.S. Guo, G.A. Ohnmacht, M.L. Parkhurst and P. Tong-On et al., 2001. Sequential 5-aza-2_-deoxycytidine-depsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY-ESO-1. J. Immunother., 24: 151-161.
PubMed - Wilson, C.A., L. Ramos, M.R. Villasenor, K.H. Anders and M.F. Press et al., 1999. Localization of human BRCA1 and its loss in high-grade, non-inherited breast carcinomas. Nat. Genet., 21: 236-240.
CrossRef - Winquist, E., J. Knox, J.P. Ayoub, L. Wood and N. Wainman et al., 2006. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: A National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Invest. New Drugs, 24: 159-167.
CrossRefPubMed - Wolffe, A.P. and M.A. Matzke, 1999. Epigenetics: Regulation through repression. Science, 286: 481-486.
CrossRef - Woodson, K., A. Flood, L. Green, J.A. Tangrea and J. Hanson et al., 2004. Loss of insulin-like growth factor-II imprinting and the presence of screen-detected colorectal adenomas in women. J. Natl. Cancer Inst., 96: 407-410.
PubMed - Wu, L.P., X. Wang, L. Li, Y. Zhao and S. Lu et al., 2008. Histone deacetylase inhibitor depsipeptide activates silenced genes through decreasing both CpG and H3K9 methylation on the promoter. Mol. Cell Biol., 28: 3219-3235.
CrossRefDirect Link - Xu, W.S., R.B. Parmigiani and P.A. Marks, 2007. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene, 26: 5541-5552.
CrossRefDirect Link - Tada, Y., M. Wada, K. Taguchi, Y. Mochida and N. Kinugawa et al., 2002. The association of death-associated protein kinase hypermethylation with early recurrence in superficial bladder cancers. Cancer Res., 62: 4048-4053.
Direct Link - Yang, A.S., M.R. Estecio, G. Garcia-Manero, H.M. Kantarjian and J.P. Issa, 2003. Comment on chromosomal instability and tumors promoted by DNA hypomethylation and Induction of tumors in nice by genomic hypomethylation. Science, 302: 1153-1153.
CrossRef - Yang, E.S. and K.L. Burnstein, 2003. Vitamin D inhibits G1 to S progression in LNCaP prostate cancer cells through p27Kip1 stabilization and Cdk2 mislocalization to the cytoplasm. J. Biol. Chem., 278: 46862-46868.
CrossRefDirect Link - Yao, X., J.F. Hu, M. Daniels, H. Shiran and X. Zhou et al., 2003. A methylated oligonucleotide inhibits IGF2 expression and enhances survival in a model of hepatocellular carcinoma. J. Clin. Invest., 111: 265-273.
Direct Link - Yegnasubramanian, S., J. Kowalski, M.L. Gonzalgo, M. Zahurak and S. Piantadosi et al., 2004. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res., 64: 1975-1986.
Direct Link - Yoo, C.B., J.C. Chuang, H. Byun, G. Egger and A.S. Yang et al., 2008. Long-term epigenetic therapy with oral zebularine has minimal side effects and prevents intestinal tumors in mice. Cancer Prev. Res., 4: 233-240.
CrossRef - Yoo, C.B. and P.A. Jones, 2006. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov., 5: 37-50.
PubMed - Yoshida, M., R. Furumai, M. Nishiyama, Y. Komatsu, N. Nishino and S. Horinouchi, 2001. Histone deacetylase as a new target for cancer chemotherapy. Cancer Chemother. Pharmacol., 48: S20-S26.
PubMed - Zhang, Y. and D. Reinberg, 2001. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev., 15: 2343-2360.
CrossRef - Zhao, X.Y., D.M. Peehl, N.M. Navone and D. Feldman, 2000. 1alpha, 25-dihydroxyvitamin D3 inhibits prostate cancer cell growth by androgendependent and androgen-independent mechanisms. Endocrinology, 141: 2548-2556.
PubMed - Zheng, Y.G., J. Wu, Z. Chen and M. Goodman, 2008. Chemical regulation of epigenetic modifications: Opportunities for new cancer therapy. Med. Res. Rev., 28: 645-687.
PubMed