
Godwin Michael Ubi
Department of Genetics and Biotechnology, Faculty of Biological Sciences, University of Calabar, P.M.B 1115, Calabar, Nigeria
LiveDNA: 234.19343
Joseph K. Ebigwai
Department of Plant and Ecological Studies, Faculty of Biological Sciences, University of Calabar, P.M.B 1115, Calabar, Nigeria
LiveDNA: 234.31468
Ndem E. Edu
Department of Genetics and Biotechnology, Faculty of Biological Sciences, University of Calabar, P.M.B 1115, Calabar, Nigeria
Paul B. Ekpo
Department of Genetics and Biotechnology, Faculty of Biological Sciences, University of Calabar, P.M.B 1115, Calabar, Nigeria
Solomon I. Ofem
Department of Microbiology, Faculty of Biological sciences, University of Calabar, P.M.B 1115, Calabar, Nigeria
Imaobong S. Essien
Department of Zoology and Environmental Biology, Faculty of Biological Sciences, University of Calabar, P.M.B 1115, Calabar, Nigeria
Trends in Bioinformatics
Year: 2021 | Volume: 14 | Issue: 1 | Page No.: 13-27







ABSTRACT
Background and Objective: In December 2019, patients diagnosed with viral pneumonia due to an unidentified microbial agent were reported in Wuhan, China. A novel coronavirus was subsequently identified as the causative pathogen, provisionally named 2019 novel coronavirus (COVID 19). The article aimed to unmask the genetic stability and nature of the viruses and validated the safety guidelines adopted for the coronaviruses currently causing the global pandemic. Materials and Methods: This study uses in silico approach and bioinformatics tools to validate the stability status and characteristics of the novel SARS-COV 2 coronaviruses by retrieving nucleotide sequences isolated and deposited in the NCBI. Results: The results have revealed that 11 out of 12 isolates studied are genetically and thermally unstable. The aliphatic index ranged from 52.98-112.07 in MT187977.1-MT127116.1, respectively. The instability index among the sequences ranged from 39.58-73.65 in MT187977.1-MT152900.1 isolate, respectively. The G-C contents range from 37.28-49.26% in MN938385.1-MT187977.1, respectively. Phylogenetically, isolates MT127116.1, MT159778.1, MT050414.1 and MT042777.1 showed the same genetic and mutation pathway with an evolutionary distance of 0.4 and 0.8 divergence times relative to the last common ancestor. Conclusion: There is a need for concerted effort in studying and understanding the genetic and thermal stability status and other characteristics of the viruses to be able to find a suitable therapy and drug design for the pandemic and biosecurity of humans against the virus now and in the future.
PDF Abstract XML References Citation
Copyright: © 2021. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Godwin Michael Ubi, Joseph K. Ebigwai, Ndem E. Edu, Paul B. Ekpo, Solomon I. Ofem and Imaobong S. Essien, 2021. Validation of Isolate Stability Status and in silico Characterization of SARS-COV 2 Partial CdsRdRP Gene Sequences. Trends in Bioinformatics, 14: 13-27.
DOI: 10.3923/tb.2021.13.27
URL: https://scialert.net/abstract/?doi=tb.2021.13.27
DOI: 10.3923/tb.2021.13.27
URL: https://scialert.net/abstract/?doi=tb.2021.13.27
INTRODUCTION
Viruses of the family Coronaviridae possess a single strand, positive-sense RNA genome ranging from 26-32 kbp sequence length1. Coronaviruses have been identified in several avian hosts as well as in various mammals, including camels, bats, porcupine, mice, dogs and cats2.
The coronaviruses are organisms with the only RNA often referred to as genetic RNA. The genetic RNA of the coronaviruses is self-replicating been able to produce its replica using the RNA dependent RNA polymerase synthesis3. The coronavirus functions directly as messengers RNA which in conjunction with the ribosomal apparatus of its host can direct the synthesis of both the RNA polymerase enzyme required for RNA replication and the production of the viral coat4. Thus, with the mediation of the RdR polymerase and based on the standard base-pairing principle, the virus RNA serves as a template in the synthesis of a complementary RNA chain and hence the synthesis of a double-stranded structure5 which with the novel mammalian coronaviruses are now regularly and easily identified.
Molecular genetics has made it possible for the easy identification of the viruses, for example, an HKU2-related coronavirus of bat origin was found to be responsible for a fatal acute diarrhoea syndrome in pigs in 20186.
Among the several coronaviruses that are pathogenic to humans, most are associated with mild clinical symptoms, with two notable exceptions: Severe Acute Respiratory Syndrome (SARS) coronavirus (SARS-CoV), a novel beta coronavirus that emerged in Guangdong, Southern China, in November, 20027 which resulted in more than 8000 human infections and 774 deaths in 37 countries during 2002-03 and the Middle East respiratory syndrome (MERS) coronavirus (MERS-CoV), which was first detected in Saudi Arabia in 2012 and was responsible for 2494 laboratory-confirmed cases of infection and 858 fatalities since September, 2012, including 38 deaths following a single introduction into South Korea8.
In late December, 2019, patients diagnosed with viral pneumonia due to an unidentified microbial agent were reported in Wuhan, China9. A novel coronavirus was subsequently identified as the causative pathogen, provisionally named 2019 novel coronavirus (2019-nCoV).
As of May, 16, 2020, confirmed cases had reached an alarming 4,642,506 with a global death rate of 308, 866 cases of 2019-nCoV infections globally, most of which involved people living in or visiting endemic regions and thus increasing the human-to-human transmission9.
Besides, 2019-nCoV has now been reported in almost 212 countries of the world including Italy, Malaysia, Belgium, Australia, Nigeria, Portugal, Germany, USA, France, Brazil, Egypt, South Africa, Canada, USA, UK, Russia, Israel, Turkey, Iran, Vietnam, China and Spain10. Infections of medical workers and family clusters have also been reported and human-to-human transmission confirmed in most of these endemic regions. Most of the infected patients had a high fever, dry cough, tiredness, chills, chest pain, shortness of breath, loss of taste and smell and dyspnea, with chest radiographs revealing invasive lesions in both lungs11-13.
The report of the epidemiological data of positively infected inpatient, from many hospitals in different endemic regions, diagnosed with viral pneumonia of unidentified cause were sequenced using next-generation sequencing of the bronchoalveolar lavage fluid samples and cultured isolates from these patients revealed the isolate as 2019-nCoV14.
The description of the genomic characterization of genomes of this novel virus will provide important information on the origins and cell-binding processes of the viruses, thus helping to chat a therapeutic measure for the viral disease15.
This in silico studies, however, was borne out of the desire to find answers to questions raised in Nigeria concerning the various preventive measures been spelt out as peddled rumours towards the effective prevention and control of the pandemic virus. Some of the peddled rumours such as the virus are thermophobic and will not thrive in tropical climes, the virus is acidic thus the use of alkaline water will be effective, the virus cannot survive for long period outside its host cells, the virus is of a large size (Micron) thus it is not airborne and hence the use of gas mask, the virus is genetically unstable and easily denature by heat above 26-27°C and virus is hydrophilic and thrive under low relative humidity16,17.
Thus it was deemed necessary to used sequences from isolated coronaviruses from different selected endemic regions including Nigeria to ascertain the Physico-chemical characteristics of the coronavirus using silicon and bioinformatics tools and also to determine whether or not, the conclusion that the coronaviruses are thermally and genetically unstable is factual or unfounded.
It is also therefore necessary for humanity to know the characteristics of the common humanity enemy called SARS-COV 2 coronaviruses. Good knowledge and understanding of the attributes/characteristics of the coronaviruses are desired to help researchers and radomedics to understand the features of the common enemy and develop strategies/approaches and designs aimed at a specific target of the coronaviruses, to bring a lasting solution to the pandemic.
This study aimed to unmask the genetic stability and nature of the viruses and validated the safety guidelines adopted for the coronaviruses currently causing the global pandemic.
MATERIALS AND METHODS
Study area: The study was carried out in the genomic/bioinformatics and molecular biology laboratory of the Department of Genetics and Biotechnology, Faculty of Biological Sciences, University of Calabar, Calabar Nigeria from March-June, 2020.
Retrieval of nucleotides and amino acid sequences: Nucleotides sequences of the SARS-COV 2 S genes were retrieved from the database of the human genome hoisted by the National Centre for Biotechnology Information (NCBI): The COVID-19 gene sequences were retrieved using the FASTA format with basic alignment search tool engine. The accession numbers for the SARS-COV 2 S gene sequences from different endemic locations were recorded after retrieval, tabulated and used for the study. The partial CDs sequences of SARS-COV 2 were retrieved and used for the study because some of the endemic regions as at the date of retrieval of sequences from the NCBI database were yet to sequence the whole genome complete sequence for the polyprotein virus thereby limiting our usage of the complete sequence for the study. It was also prompted by the fact that Nigeria as at the time also deposited the partial sequences hence our choice of the partial cdsRdRP region for the study. Moreso, the whole complete genome sequences from the isolates were not used for the preliminary in silicon studies because of the anticipated large volume of introns (non-coding regions of the gene).
Hence, nucleotide sequences were downloaded for RdRP region partial CDs gene sequences deposited from different countries into the NCBI with the following accession numbers MT0088022.1, MT042777.1, MT050414.1, MT066157.1, MT172668.1, MT152900.1, MT127116.1, MT159778.1, MT187977.1, MN938385.1, LC522350.1 and MN970003.1.
Determination of physicochemical properties of the novel coronaviruses genes sequences: The proteomic, protparam options of the Expasy (Expert Protein Analysis System) online interactive software program was used to determine the physicochemical properties of the novel SARS-COV 2 (COVID-19) S gene sequences using the retrieved nucleotide and translated amino acid sequence of the different gene sequences. This analysis was used to determine the molecular weight, atomic composition of the gene sequences, total negative charged amino acid and positively charged amino acid residues, theoretical isoelectric point pH (pl), extinction coefficient, instability index, extinction coefficient and grand hydropathicity of the novel SARS-COV 2 gene sequences.
Determination of guanine-cytosine contents start and end codons and coding regions of SARS-COV 2 sequences: The interactive online program GENSCAN was used to determine the percentage of guanine and cytosine contents in the nucleotide sequences of the SARS-COV 2 among the selected endemic regions.
Determination of phylogenetic relationship among the novel SARS-COV 2 gene sequences: The molecular evolution and Genetic analysis (MEGA X.0) software were used to determine the evolutionary relationship existing between the SARS-COV 2 genes sequences obtained from the different endemic regions. The retrieved sequences were aligned using the CLUSTALW option of multiple sequence alignments of MEGA X.0.
The phylogenetic tree was constructed using an unweighted group paired mean averages (UPGMA) option at 1000 boots trap of the retrieved amino acid sequence of the novel SARS-COV 2 genes. The time of divergence of the novel SARS-COV 2 gene sequences were also inferred. The MEGA X software was used to align the sequences and subject them to phylogenetic analysis for substitution model selecting, evolutionary distance estimation, phylogeny inference, substation rate and pattern estimation, the test of natural selection and ancestral sequence inference for the selected sequences.
Prediction of secondary and tertiary protein folding RNA structure of coronavirus sequences: The secondary protein folding structures of the FASTA formatted coronavirus sequences were predicted using the online interactive program NSOPMA for the determination of dominance of the folded secondary structures by alpha-helices, beta turns, extended strands and random coils. Another online interactive program, the phyre and phyre (protein homology Y recognition engine) which is based on the canonical amino acid sequence obtained from the NCBI database18 was used for the determination of the tertiary protein folding structures for the coronavirus sequences.
RESULTS AND DISCUSSION
Physical characteristics of the selected novel SARS-COV 2 coronavirus sequences
Molecular weights/sizes of the novel SARS-COV 2 coronaviruses: The preliminary in silicon evaluation for the determination of Physico-chemical characteristics of the retrieved partial cds nucleotide and translated protein sequences isolated from the RNA-dependent-RNA polymerase (RdRP) region of the novel coronavirus strains from 12 selected endemic regions of the world showed that the number of translated amino acid from the retrieved nucleotides sequence using the MEGA X.0 translation option varied from 51-227 KDA in LC522350.1-MT172668.1 isolates, respectively in Table 1. Three nucleotides make up an amino acid codon. The molecular weight of the SARS-COV 2 RdRP region gene sequences also ranged between 5597.51-25,734.22 g mol–1 in LC522350.1-MT172668.1 isolate, respectively. The molecular weights varied among the isolates (Table 1) and thus exhibit differences in characteristics.
The coronavirus isolates also showed variability in the number of amino acids contained in their molecules. The number of amino acids was higher 227 in MT172668.1 isolate and at least 51 in LC522350.1 isolates.
Coronaviruses are single-stranded RNA genome with genomic molecular sizes ranging from 26000-32000 bp in length. The genome of the viruses has approximately 35 functional proteins/genes within the genome. Each gene occupying approximately between 200-3000 bp. Most of the sequences in the genome are introns implying the non-coding region of the genomes18-20. The molecular weight of the isolates plays a critical role in the dimensional structures of the coronaviruses as they determine the site for chemical formula derived descriptors during drug design, lead and optimization.
Total number of atoms of the novel SARS-COV 2 coronaviruses: The total number of atoms possessed by each of SARS-COV 2 isolates determines it strength and stability. The more atoms found in an isolate, the more stable the isolate as more heat and other compounds are needed and required to destroy such isolate. The total number of atoms varies directly with the molecular weight and number of amino acids of the isolates. Isolates with higher molecular weight are those with a higher number of atoms as shown in Table 1.
It was gathered from the study that the total number of atoms from the isolates showed that the least was from LC522350.1 isolate had 782 atoms while MT172668.1 isolate had the highest number of 3573 atoms (Table 1).
Amino acid side chain constituents of the novel SARS-COV 2 coronaviruses: The amino acids side chain constituent of the protein coat of the viruses plays a very significant role in the reactivity of their antigen with the antibody of the host immune system. The amino acids side chain is the fulcrum upon which the immunochemistry of drugs, heptans and antibodies revolves. The heavy and light amino acid side chains take part in drug delivery reactions to target sites. The design of drugs using the ligands based approach is highly dependent on the heavy and light amino acids side chains of the reacting species21,22. Table 2 presents the results of dominant side-chain amino acids for the coronaviruses and their percentage bioavailability for the reaction. Alanine side-chain occupied 8.0% in MT042777.1 isolate, Valine side-chain occupied 8.6% in MT127116.1 isolate and isoleucine side chain occupied 6.2% of MN938385.1 isolate while Leucine occupied 17.1% of the side chain amino acids in MT0088022.1 isolate (Table 2).
Aliphatic (side chain) index of the novel SARS-COV 2 coronaviruses: Aliphatic index of the virus protein sequence is the relative volume occupied by the aliphatic side chains including alanine, valine, isoleucine and leucine amino acids respectively. The measure of the aliphatic index of a protein is a positive factor in increasing the thermostability of the globular proteins of the viruses. Globular protein stability varied among the coronavirus sequences and ranged from 52.98-112.07 in MT187977.1-MT127116.1 isolate, respectively. Globular protein stability decreases with increasing index volume occupied by the aliphatic side chain. As the volume occupied by the aliphatic side chain consisting primarily of the amino acids, alanine, valine, isoleucine and leucine increases, the thermostability of the virus decreases as the amino acids are synthesized with time23. Hence, the results in Table 2 shows that the RdRP region sequence from MT187977.1 isolate was more genetically and thermally stable while that from Vietnam MT127116.1 isolate was the least unstable.
Instability index of the novel SARS-COV 2 coronaviruses: The instability index is a characteristic that determines the genetic and thermal stability of the sequences. An RNA sequence is said to be thermally and genetically stable if its instability index is below 40 and scientifically considered thermally and genetically unstable if its instability index is 40 and above. Hence from the results, it implies that only the isolate sequence from MT187977.1 isolate was thermally and genetically stable while all other 11 sequences were unstable. The results as presented in Table 3 further revealed that the instability index among the sequences ranged from 39.58-73.65 in MT187977.1-MT152900.1 isolate sequences, respectively.
| Table 1: Some physico-chemical parameters of SARS-COV 2 coronavirus sequences | |||||
No. of amino | Molecular | Total No. of | Total No. of a negatively | Total No. of a positively | |
| Accession no. | acids | weight | atoms | charged residue | charged residue |
| MT0088022.1 | 99 | 10998.77 | 156.1 | 8 | 12 |
| MT042777.1 | 87 | 9643.92 | 1328 | 7 | 9 |
| MT050414.1 | 167 | 18918.91 | 2636 | 14 | 17 |
| MT066157.1 | 82 | 9654.11 | 1323 | 6 | 9 |
| MT172668.1 | 227 | 25734.22 | 3573 | 5 | 23 |
| MT152900.1 | 92 | 10858.55 | 1530 | 2 | 21 |
| MT127116.1 | 140 | 15806.59 | 2240 | 9 | 13 |
| MT159778.1 | 145 | 16289.82 | 2266 | 11 | 14 |
| MT187977.1 | 131 | 14999.90 | 2083 | 10 | 18 |
| MN938385.1 | 81 | 8905.49 | 1259 | 1 | 11 |
| LC522350.1 | 51 | 5597.51 | 782 | 0 | 8 |
| MN970003.1 | 82 | 9655.11 | 1323 | 6 | 9 |
| Table 2: Amino acid side chain composition and aliphatic index of coronavirus sequences of some endemic regions | |||||
| Accession no. | Alanine (%) | Valine (%) | Isoleucine (%) | Leucine (%) | Aliphatic index |
| MT0088022.1 | 6.1 | 5.1 | 5.1 | 17.2 | 102.37 |
| MT042777.1 | 8 | 6.9 | 1.1 | 11.5 | 77.36 |
| MT050414.1 | 5.4 | 7.2 | 3.0 | 14.4 | 93.93 |
| MT066157.1 | 4.9 | 6.1 | 2.4 | 9.8 | 70.12 |
| MT172668.1 | 3.5 | 6.2 | 5.3 | 11.5 | 86.70 |
| MT152900.1 | 7.6 | 5.4 | 2.2 | 9.8 | 70.00 |
| MT127116.1 | 6.4 | 8.6 | 5.0 | 15.7 | 112.07 |
| MT159778.1 | 6.2 | 7.6 | 2.8 | 14.5 | 95.45 |
| MT187977.1 | 7.6 | 2.3 | 4.6 | 5.3 | 52.98 |
| MN938385.1 | 6.2 | 4.9 | 6.2 | 9.9 | 83.09 |
| LC522350.1 | 7.8 | 3.9 | 5.9 | 3.9 | 57.54 |
| MN970003.1 | 4.9 | 6.1 | 2.4 | 9.8 | 70.12 |
| Table 3: Instability index, guanine-cytosine contents and in vitro mammalian reticulocytes estimated half-life of coronaviruses | |||
| Accession no. | Instability index | G-C content (%) | Estimated half-life |
| MT0088022.1 | 62.26 | 45.39 | 1.9 |
| MT042777.1 | 46.24 | 38.78 | 4.4 |
| MT050414.1 | 51.63 | 38.08 | 2.0 |
| MT066157.1 | 40.51 | 38.62 | 1.4 |
| MT172668.1 | 54.39 | 37.53 | 1.9 |
| MT152900.1 | 73.65 | 48.48 | 1.9 |
| MT127116.1 | 46.37 | 38.34 | 1.3 |
| MT159778.1 | 49.67 | 38.51 | 1.2 |
| MT187977.1 | 39.58 | 49.26 | 7.2 |
| MN938385.1 | 49.18 | 37.28 | 1.0 |
| LC522350.1 | 40.6 | 38.46 | 1.0 |
| MN970003.1 | 40.51 | 38.62 | 1.4 |
With a wide spread unfavourable instability index among the evaluated COVID 19 coronaviruses, it implies that there are easily denatured by heat (high temperature) and other elements of weather. This thus confirms the assertion that the coronaviruses do not thrive in a tropical climate with high temperatures and high humidity24.
Guanine-cytosine contents of the novel SARS-COV 2 coronaviruses: The guanine-cytosine (G-C) content of a gene sequence is a measure of the genetic and thermal stability of the gene sequences. The guanine-cytosine linkage in the double-strand molecule shows that guanine and cytosine (G = C) are amino acids that are held by triple hydrogen bond in the double-stranded RNA molecule while the adenine and uracil (Thymine) amino acids are held together by a double hydrogen bond. Hence the triple hydrogen bonds which are covalently bonded require more thermal (heat) energy inputs for dissociation compared to the thermal energy required for dissociation of the double bond (A = T). Hence a nucleotide sequence with more guanine and cytosine will have more triple bonds and thus will be genetically and thermally stable25-27. However, based on the guanine and cytosine content of a gene sequence, a gene is said to be thermally and genetically stable if its G-C content is 49% and above and below which 0-48% it is said to be genetically and thermally unstable. In Table 3, the G-C contents of the RNA sequences of the isolates range between 37.28-49.26% in MN938385.1-MT187977.1 isolate, respectively indicating that only the sequences from MT187977.1 isolate was genetically and thermally stable while others were unstable and thus easily prone to gene mutations27.
Half-life of novel SARS-COV 2 coronaviruses in human reticulocyte cells: The half-life is a prediction of the time it takes for half of the amount of protein in a cell to disappear after its synthesis in the cell. It relies on the N-terminal amino acid (side chain) or N-end rule which relates the half-life of a protein to the identity of its N-terminal amino acid residue. Thus, the N-terminal amino acids originated from the observation that the identity of the N-terminal residue (aliphatic side chain) of a protein plays an important role in determining the protein stability in vivo28. The half-life of the isolates evaluated in Table 3 ranged from 1.0 h in MN938385.1 and LC522350.1 isolates to 7.2 h in MT187977.1 isolate. Thermal and genetic stability of isolates is associated with N-Terminal amino acid which is a measure of the isolate stability. The stability and mutation rate of the isolate sequences increases with an increase in half-life. The MT187977.1 isolate showed the highest half-life of 7.2 h and hence its thermal and genetic stability status (Table 3).
Essential chemical characteristics of the novel SARS-COV 2 coronaviruses
Grand hydropathicity of the novel SARS-COV 2 coronaviruses: The hydropathicity character of an isolate sequence shows the free energy of transfer G (Kcal mol–1) from the solvent to solute interface. It involves the transfer of unfolding protein chains from water to the bilipid layer interface and the transfer of unfolded chains to organic solvent29. This characteristic explains the use of alcohol-based sanitisers for the prevention of coronaviruses.
The grand hydropathicity which is an expression of the hydrophobicity and hydrophilicity character of the virus protein coat was determined for the various SARS-COV 2 virus isolates and the results as presented in Table 4 revealed that eight of the twelve isolates expressed hydrophobic character while only four expressed hydrophilic character. Hydrophobicity characteristics of the isolates varied from-0.053-0.777 in MT008022.1-MT187977.1 and MT152900.1 isolates, respectively. Table 4 also revealed that hydrophilicity characteristics of the isolates ranged from 0.05-0.344 in MT050414.1-MT127116.1 isolate, respectively in Table 4.
The grand hydropathicity value compares the most abundant protein in the extracellular matrix. Hydropathicity illustrates the therapeutic use of water as determined by the hydrophobicity scale30. This also shows the extent to which the water hand washing sanitizing hygiene adopted is effective for the prevention of the virus spread. Hydrophobicity scales are also used to predict the preservation of the genetic code for the viruses31. The results are in tandem with the reports of31who measured the free energy of unfolding protein chain from isolates and found that increase in genetic and thermal stability of the protein structure is directly proportional to increase in hydrophobicity of the protein up to a certain size limit32. Hence the conclusion that the protein structure stability is measured by its hydropathicity character.
Theoretical isoelectric point (PL) of the novel SARS-COV 2 coronaviruses: The theoretical isoelectric point (pl) represents the pH of a solution at which the net charge of the isolates protein coat becomes zero23,33. In solutions in which the actual pH is above the theoretical isoelectric point, the surface of the isolated protein would predominantly be negatively charged and therefore like-charged molecules, would exhibit or show repulsive forces because it carries no net electrical charge and thus is electrically neutral24,33. The theoretical isoelectric point is based on the primary protein structure which is the primary sequence that is unlikely to match the actual pH due to some charged side chains forming a salt bridge25,34. The theoretical isoelectric point value affects the solubility of the protein molecule at this given pH, by conferring minimum solubility in water and salt due to precipitation of the salt in solution. This accounts for the use of alcohol-based sanitisers in dissolving the protein coat of the novel coronaviruses. Thus a specific theoretical pl for a target protein sequence can be used to model the process from which compounds used for the purification of the protein can be developed34. The theoretical isoelectric (pl) point varies from 8.33 in an isolated MT159778.1 to 11.95 obtained from MT152900.1 isolate in Table 4.
Extinction/attenuation coefficients of the novel SARS-COV 2 coronaviruses: The extinction coefficient (EC) is a characteristic that determines how strongly a species absorbs or reflects radiations or light at a particular wavelength. It is an intrinsic property of the isolates that is dependent on the atomic, chemical and protein structural composition of the isolate sequences. Hence, when high ultraviolet light is incident on an object containing the coronavirus, the extinction coefficient characteristics make it possible for the viral protein coat to be broken down completely or disintegrate into extinction. This characteristic was low for all isolates except for the MT187977.1 isolate as shown in the results in Table 4.
The light or radiation absorption and reflection potentials (extinction coefficient) towards the disintegration of the protein coat of the isolates vary from 9190-54,430 m mol–1 in MT042777.1-MT187977.1 isolate, respectively (Table 4). The attenuation coefficient is the ability of the coronaviruses to be weakened in terms of their virulence after light absorption due to genetic and thermal disintegration. This coefficient varied among the mutant strains of the coronaviruses (Table 4). This singular characteristic of the SARS-COV 2 coronaviruses occasioned by the fast rate of mutation of the coronaviruses has made the production and development of vaccines (an attenuated form of the coronavirus) difficult as the mutant strains possess variable attenuation coefficients with which resurgence is possible after attenuation.
Total number of negatively charged amino acids residues of the novel SARS-COV 2 coronaviruses: The total number of negatively charged amino acid residues is the total of Aspartic amino acids and Glutamine amino acids in the coronavirus protein coat. This characteristic of the viral protein coat determines its reactivity with the solvent. Highly negatively charged residues will thrive and react to acidic media26,28,33.
The total number of negatively charged amino acid residues from the SARS-COV 2 isolates were also determined and presented in Table 5. It shows that the theLC522350.1 isolate did not have any negatively charged amino acid residue within the coding region while the isolate from MN938385.1 had only one negatively charged amino acid residue in the coding region. The isolate from MT050414.1 had the highest number of negatively charged amino acid residues of 14 in the coding region. All the isolate sequences evaluated in this report contain a lower number of total negatively charged Aspartic and Glutamine amino acid residues. The use of alkaline water here does not affect the viruses as like-charges will repel.
Total number of positively charged amino acids residues of the novel SARS-COV 2 coronaviruses: The total number of positively charged amino acid residues is the total of Arginine amino acids and Lysine amino acids in the virus protein coat. This characteristic of the viral protein coat determines its reactivity with various solvents. Highly positively charged residues will thrive on alkaline media27,29,33.
Table 5 further showed that the total number of positively charged amino acid residue in the coding region of the retrieved and translated sequences varied from 8 in LC522350.1 isolate to as high as 23 in MT172668.1 isolate. All the isolate sequences evaluated contains a higher number of total positively charged Arginine and Lysine amino acid residues. The use of alkaline water greatly affects these classes of coronaviruses due to the reaction of the positively charged amino acid residues and the alkaline water. Thus, the prescription on the use of alkaline water as a means of preventing the virus spread in some quarters.
Coding regions of the novel SARS-COV 2 coronaviruses: The exons or coding regions reveals the portion of the virus genome that contains the Spike protein gene that codes for the protein that contains the inoculum or infective entity. The results of preliminary in silicon evaluation for the determination of the coding region of the SARS-COV 2 for the retrieved partial cds nucleotide and translated protein sequences isolated from the RNA-dependent-RNA polymerase (RdRP) region of the novel coronavirus strains from 12 selected endemic regions of the world are presented in Table 6. It showed that the exon or coding regions data were not available for MT0088022.1, MT066157.1, MN938385.1, LC522350.1 and MN970003.1 isolates. However, the coding region data range from 9-227 bps in MT042777.1 Isolate, 11-424 bps in MT050414.1 isolate, 9-324 bps in MT172668.1 isolate, 40-309 bps in MT152900.1 isolate, 16-234 bps in MT127116.1 isolate, 9-275 bps from MT159778.1 isolate and 50-173 bps in MT187977.1 isolate (Table 5).
Biological characteristics of the novel SARS-COV 2 coronaviruses
Protein coat structure of the novel SARs-COV 2 coronaviruses: The protein coat structures of the viruses can enable the protein engineering of the fat which occupies the outer layer of the virus protein coat and determine the substrate specificity, double bond positioning (regioselectivity) and reaction outcome of the coronaviruses28,31,33. Information on the protein coat characteristics can also provide the potentials to improve effectiveness in existing reaction mechanisms and even to engineer enzymes that can undertake or undergo reactions at novel positions in the carbon side chains. Knowledge of the protein folding structures can open up the possibility of synthesizing novel fatty acids with multiple functional groups there by paving the way for designer lipids that currently lie beyond the reach of most of the available metabolic or protein engineering strategies, thus initiating a pathway for therapeutic enhancement and discovery29,34.
The development and use of fatty acids desaturases are prime targets in protein engineering because of their central and well-defined role in the modification of aliphatic side chains (alanine, valine, isoleucine and leucine) within the cell.
| Table 4: Extinction coefficient, theoretical isoelectric point and grand hydropathicity of RdRP region of coronavirus sequences | ||||
| Accession no. | Nucleotide bps | Extinction coefficient | Theoretical isoelectric point (pl) | Grand hydropathicity |
| MT0088022.1 | 322 | 12740 | 9.04 | -0.053 |
| MT042777.1 | 294 | 9190 | 8.37 | -0.255 |
| MT050414.1 | 562 | 18170 | 8.28 | 0.05 |
| MT066157.1 | 250 | 17795 | 8.57 | -0.106 |
| MT172668.1 | 810 | 37815 | 9.26 | 0.181 |
| MT152900.1 | 322 | 37595 | 11.95 | -0.777 |
| MT127116.1 | 459 | 20315 | 8.74 | 0.344 |
| MT159778.1 | 483 | 15065 | 8.33 | 0.106 |
| MT187977.1 | 406 | 54430 | 9.83 | -0.777 |
| MN938385.1 | 287 | 12740 | 10.63 | -0.042 |
| LC522350.1 | 182 | 12615 | 10.57 | -0.292 |
| MN970003.1 | 290 | 17795 | 8.57 | -0.106 |
| Table 5: Start and end codon and coding (exon) region of RdRP region of coronavirus sequences | ||||
| Accession no. | RdRP length (bps) | Start codon | End codon | Coding region |
| MT0088022.1 | 322 | NA | NA | NA |
| MT042777.1 | 294 | 9 | 227 | 9-227 |
| MT050414.1 | 562 | 11 | 424 | 11-424 |
| MT066157.1 | 250 | NA | NA | NA |
| MT172668.1 | 810 | 224 | 712 | 9-324 |
| MT152900.1 | 322 | 17 | 287 | 40-309 |
| MT127116.1 | 459 | 69 | 441 | 16-234 |
| MT159778.1 | 483 | 22 | 414 | 9-275 |
| MT187977.1 | 406 | 19 | 282 | 50-173 |
| MN938385.1 | 287 | NA | NA | NA |
| LC522350.1 | 182 | NA | NA | NA |
| MN970003.1 | 290 | NA | NA | NA |
| Table 6: Secondary protein folding characteristics of coronavirus sequences of some endemic regions of the world | ||||
| Accession no. | Alpha helix (%) | Extended strand (%) | Random coil (%) | Beta turns (%) |
| MT0088022.1 | 25.25 | 17.17 | 51.52 | 6.06 |
| MT042777.1 | 56.32 | 13.76 | 20.69 | 9.20 |
| MT050414.1 | 50.90 | 13.72 | 24.55 | 10.78 |
| MT066157.1 | 31.71 | 37.80 | 23.17 | 7.32 |
| MT172668.1 | 39.21 | 25.58 | 25.55 | 9.69 |
| MT152900.1 | 38.02 | 9.78 | 41.30 | 10.87 |
| MT127116.1 | 63.57 | 10.00 | 20.00 | 6.43 |
| MT159778.1 | 49.66 | 19.31 | 22.07 | 8.79 |
| MT187977.1 | 1.53 | 22.14 | 67.94 | 8.40 |
| MN938385.1 | 51.85 | 12.38 | 30.86 | 4.94 |
| LC522350.1 | 25.49 | 29.41 | 35.29 | 9.8 |
| MN970003.1 | 31.71 | 36.59 | 34.39 | 7.32 |
This may further provide an insight into the structural determinants that govern the regioselectivity within the acyl chain of the fatty acid substrate that makes up the viral protein coat30,34. Knowledge of the structure of the protein of the coronaviruses can be very useful in structure-based drug design and ligand-based drug design and discovery.
Primary protein structures of the novel SARS-COV 2 coronaviruses: The primary protein structure is the particular sequence of amino acids found in the coronavirus protein coat and is determined by the covalent peptide bonding between the amino acids. The primary protein structures for the coronavirus are the basic nucleotide and amino acid sequences. This structure plays a significant role in chemical formula derived descriptors for computer-aided drug design for the coronavirus disease31,33,34.
Secondary protein folding structures of COVID-19 coronaviruses: The secondary folding structure of a protein is the regular repeating organization of the polypeptide heavy and light chains. This characteristic of the coronaviruses determines to a large extent, the reactivity of the viral protein coat, the thermal as well as the genetic stability of the spike protein gene of the coronaviruses. The dominant sheet of the secondary structure determines their genetic strength and stability32,34.
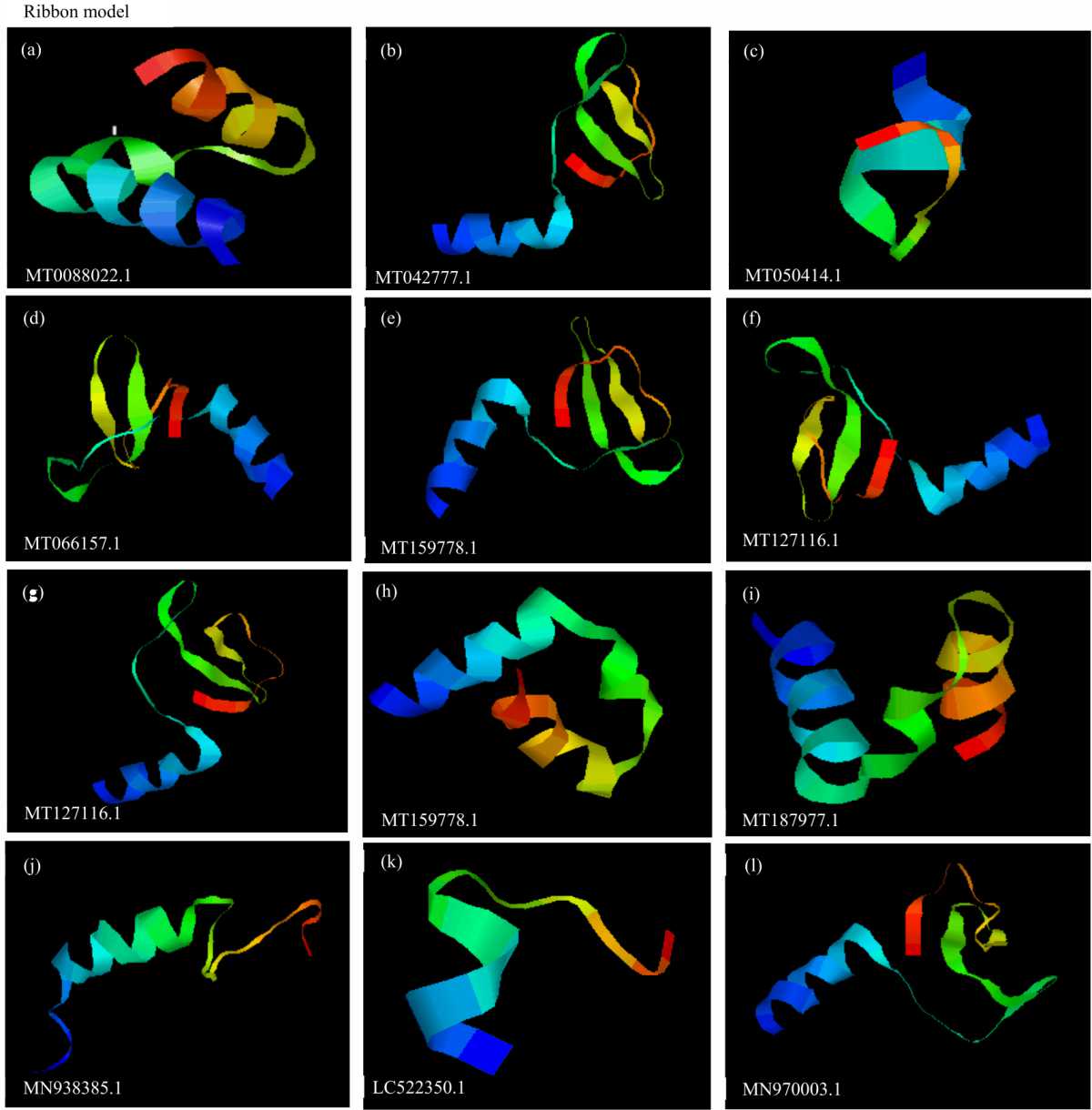 |
| Fig. 1(a-l): | 3-D tertiary protein folding (ribbons and other types) structures of partial cdsRdRP region of novel coronaviruses sequences |
Secondary structures dominated by alpha-helices (building blocks) are more stable, closely followed by those dominated by random coils and then the extended strands. Some also show the presence of beta turns regulatory elements in their molecule.
Table 6 shows that the secondary protein folding structure of the RdRP region RNA sequence was dominated by Alpha helices for the sequences from MT042777.1 isolate (56.32%), MT050414.1 isolate (50.90%), MT172668.1 isolate (39.21%), MT127116.1 isolate (63.57%), MT159778.1 isolate (49.66%) and MN938385.1 isolate (51.85%). The secondary protein folding structure of the RdRP region RNA sequences was dominated by extended strand for sequences from MT066157.1 isolate (37.80%) and MN970003.1 isolate (36.59%) while the random coil was the dominant secondary folding protein structure sheet for sequences obtained from LC522350.1 isolate (35.29%), MT187977.1 isolate (67.94%), MT152900.1 isolate (41.30%) and MT008022.1 isolate (51.52%) in Table 6.
Tertiary protein folding structures of COVID-19 coronavirus: Tertiary protein folding structures are simply the more compact structure in which the helical and non-helical regions of a polypeptide chain are folded back on themselves which occurs in specific patterns thereby conferring certain characteristics (3-D) properties on the protein. The 3-D tertiary protein folding structure can be model and optimize to discover drugs for small molecules (ligands)33,34. With the development of the structural 3-D model of the coding regions of the coronaviruses as shown in Fig. 1(a-l), it is very possible and important in the development of prophylactics and therapeutics for the dreaded disease using the structure-based drug design and ligand-based drug design by adopting the computer-aided drug design (CADD, SBDD and LBDD) approaches. The 3-D tertiary protein structures provide reliable descriptors and techniques for comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) in drug design, lead and optimization34.
Preliminary in silico determination of Tertiary folding protein coat structure for the NCBI retrieved partial cds translated protein sequences obtained from the RNA-dependent-RNA polymerase (RdRP) region of the novel coronavirus strains from selected endemic regions of the world are presented in Fig. 1(a-l).
Motif and reaction site of the novel SARS-COV 2 coronaviruses: Biosynthetic processes of the RNA replication, transcription and synthesis take place in sites like N-glycosylation site, C-phosphorylation site, N-myristoylation site, ADP ribosylation factor and amidation motif sites (Table 7) hat are similar among the mutant strains of coronaviruses. The motifs of the isolates as presented in Table 7 revealed that the isolates share important characteristics in terms of their reaction site, protein targets and biochemical synthesis. The results show that the SARS-COV 2 viruses have four main protein types common to all the isolates which include the spike (S-protein), Nucleocapsid (N-protein), Envelope (E-protein) and the membrane (M-protein). Knowledge of the motifs of the coding region of the coronaviruses will reveal the organization of the antibody molecule and its domain as well as the likely reaction site thereof33,34. The biosynthesis of glucose for instance among the isolates takes place in the N-glycosylation site, while the synthesis of protein takes place at the ribosylation motifs. Similarly the energy production site takes place at the ADP C-phosphorylation motifs. It is worth mentioning that even though these motifs sites are very crucial to the survival of the isolate pathogen inside its host, the isolate operates in different protein target site to achieve similar results (Table 7). Example, whereas the isolate MT042777.1 achieves glucose biosynthesis at 45-48 bp, the isolate MT05414.1 carry out glucose biosynthesis at 69-72 bp with same size of gene base pair (Table 7). These protein sites are the protein coat surface antigens through which the viruses infect their host with their toxins. It is important to note that the viruses though have in common the aforementioned protein bodies, the expression of the protein as the surface antigen site for the “peeling off” infection of host differs among the mutant strains of the coronaviruses34. This characteristic feature allows the coronaviruses to escape the immune system and phagocytosis action of the host antibodies and also attenuate vaccines used for any related respiratory tract ailment like SARS-COV 1. These attributes of the coronaviruses have also made it possible for the lowered efficacy of drugs like chloroquine previously acknowledged and used for the prophylactic and therapeutic treatment of SARS-COV 1 in 2003/4. Thus this calls for thorough research into the dominating surface antigen protein types exhibited by the current mutant SARS-COV 2 strains before the development and design of target drugs and vaccines.
Antigen epitope characteristics of the novel SARS-COV 2 coronaviruses: An epitope is that part of the virus pathogen which binds to the paratope of the host antibody. The paratope is the part of the host antibody that recognizes an antigen-binding site and is usually a small region (15-22 amino acids) of the host antibody Fv region which contains parts of the antibody heavy and light chains. The antigen receptor part of the antibody for coronaviruses is the Angiotensin-converting enzyme usually designated as (ACE-2). At this site, the host immune system produces cytokine storms including interleukins, Tumor necrotic factors that react with the viral antigen forming the epitope. Specific antibody produced by patients specific to the viral antigen offers a very unique opportunity for drug design and discovery, diagnosis and treatment.
Phylogenetic characterization of the novel SARS-COV 2 coronaviruses
Evolutionary distances of the novel SARS-COV 2 coronaviruses: Preliminary in silico evaluation for the determination of developmental relationship among the coronaviruses was carried out by inferring evolutionary pathway using the phylogenetic tree and evolutionary time of divergence for the retrieved partial cds nucleotide sequences isolated from the RNA-dependent-RNA polymerase (RdRP) region of the novel coronavirus strains from 12 endemic regions and presented in Fig. 2. The phylogenetic tree is a rooted tree indicating that the evaluated coronaviruses evolved from a common ancestor. This brings to the fore, the basic truth to clarify the “doubting Thomases” that the coronaviruses exist but in mutant forms in the various endemic regions evaluated. The molecular evolutionary and genetic analysis (MEGA) software was used to infer evolutionary distance, mutation pathway and phylogenetic (developmental) relationship among the retrieved RdRP region COVID-19 sequences. The reference sequence used for inferring the mutation pathway and evolutionary distance was the MN908947.3 Beta viruses SARS-COV 2 obtained as reference sequences deposited in the NCBI database. The reference sequence showed a percentage homology of 99.8% with most of the predicted sequences in terms of similarity and identity. The evolutionary distance and consensus of the reference sequence were1.6 indicating the most time of genetic divergence of the SARS-COV 2 gene more than other evolving ones.
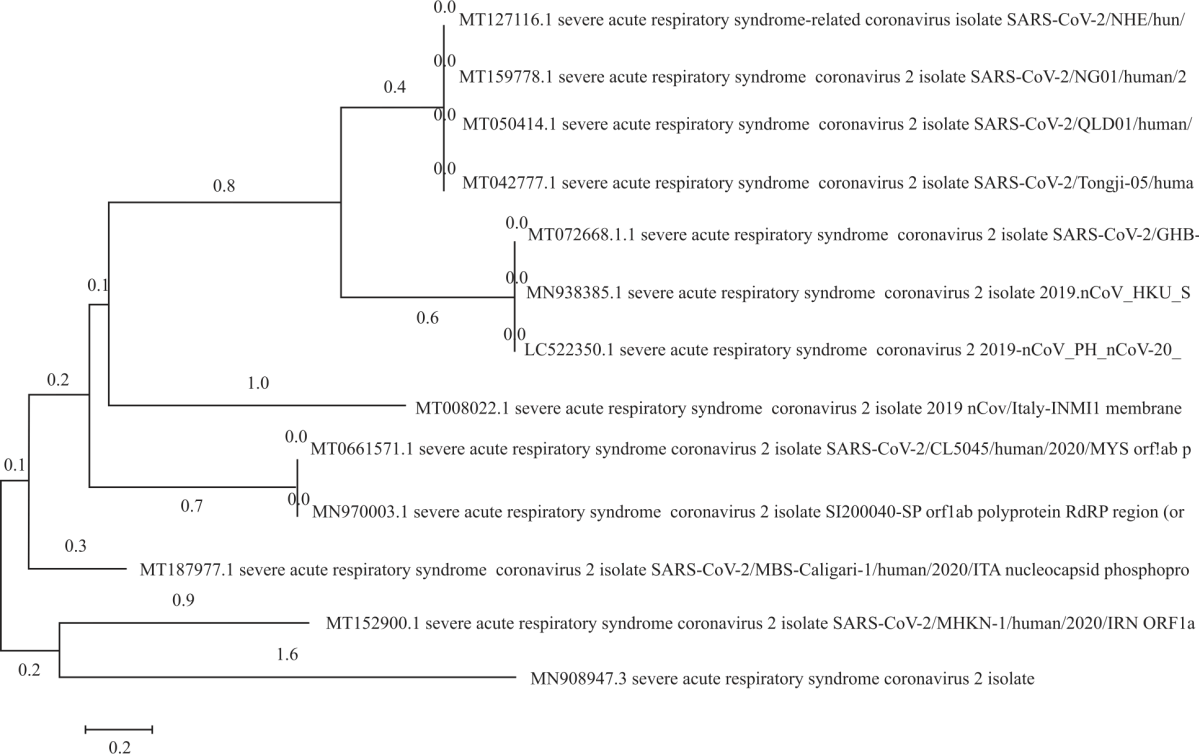 |
| Fig. 2: | Phylogenetic tree showing evolutionary distance, developmental relationship and mutation pathway among selected coronaviruses mutant strains |
| Table 7: Similarity and variations in motifs showing reaction sites and their coding regions | ||||||
| Gene bank | ASN_glycosylation | N_myristoylation | CK2_phosphorylation | Carbohydrate. | PKC_phosphorylation | cAMP&cGMP dependent- |
| acc. no. | site | site | site | MOD RES | site | phosphorylation site |
| MT0088022.1 | 27-32GSatSL | 13-16 SPvE | MOD.RES | 53-55 SqR | ||
53, 67 | 67-69SrK | |||||
| MT042777.1 | 45-48 NLSS | 30-35 GgtsSG | 45GIcANc | 47-49 SsR | ||
31-36GTssGA | MOD.RES | |||||
35-40GAttAY | 47 | |||||
59-64GNkiAD | ||||||
| MT050414.1 | 69-72 NLSS | 54-59GGtsSG | 6-9 TcIE | 6 GicNAc | 71-73SsR | |
55-60GTssGA | MOD.RES | 124-126TIR | ||||
59-64GAttAY | 6, | |||||
83-88GNkiAD | 124 | |||||
| MT066157.1 | 1-4 NTSY | 27-30 ItE | 4GIcNAc | |||
MOD.RES. 27, 79 | 55-58 RRaT | |||||
| MT172668.1 | 54-57NWTS | 50-55GAtsNW | 112-115 SsiE | 54 GIcNAc | 34-36TnR | |
76-81GLgLSM | MOD. RES | 38-40SsK | ||||
78-83GLsmSH | 112 | 56-58TsK | ||||
89-94GLtcSC | 64-66TmK | |||||
148-153GOccTF | 172-174SIK | |||||
194-196TIR | ||||||
| MT152900.1 | 42-47 GNKgAW | MOD.RES | 11-13 TrR | 12-15 RRhs | ||
11, 15 | 15-17 SvR | 50-53 RRrS | ||||
64-66 SIR | ||||||
| MT127116.1 | 88-91 NLSs | 14-19 GliINV | 25-28 TcIE | 88 GLcANc | 90-92 SsR | |
73-78GGtsSG | MOD.RES 25, 90, | |||||
74-79GTssGA | ||||||
78-83GAttAY | ||||||
102-107GNkiAD | ||||||
| MT159778.1 | 66 – 69 NLSS | 51-56 GGtsSG | 3 – 6 TcIE | 66-GIcNAc | 68-70 SsR | |
52-57GTssGA | 121-123 TIR | |||||
56-61GAttAY | 121 | |||||
80-85GNkiAD | ||||||
| MT187977.1 | 29-34 GAikTT | 70-73 SspD | MOD. RES | 1-3 TpK | 80-83 RRat | |
63-68GVpiNT | 71-74 SspD | 1, 4, 15, 70, 71, 83, 97 | 4-6 SaK | |||
112-117GLpyGA | 107-110TqpE | 15-17 TIR | ||||
83-85 TrP | ||||||
97-99 SpR | ||||||
| MN938385.1 | 61-64 NWTS | 3-8 GSrcTS | 61 GLcANc | 41-43 TnR | ||
57-62 GAtsNW | MOD.RES | 45-47 SsK | ||||
41, 45,71 | 63-65TsK | |||||
71-73TmK | ||||||
| LC522350.1 | 31-34 NWTS | 27-32 GAtsNW | 31 GLcANc | 11-13 TnR | ||
MOD. RES | 15-17 SsK | |||||
11, 15, 33,41 | 33-35 TsK | |||||
41-43 TmK | ||||||
| Ck2: Casein kinase ii phosphorylation site, PKC: Protein kinase C phosphorylation site, CAMP: CAMP and cGMP-dependent protein kinase phosphorylation site, MOD RES: Phosphothreonine and phosphoserine conditions and GlcNAc: Asparagine | ||||||
The closest relative to the Beta virus reference sequence was the partial cdsRdRP region of SARS-COV 2 isolate from MT152900.1 and also the analysis obtained with MT152900.1 isolate showed an evolutionary distance and mutation pathway of 0.9 and group consensus of 0.2 relative to the last common ancestor and reference sequence (Fig. 2).
Time of genetic divergence and group consensus of the novel SARS-COV 2 coronaviruses: The phylogenetic tree presented (Fig. 2) revealed that the SARS-COV 2 sequences retrieved from the NCBI database from partial cdsRdRP region of SARS-COV 2 isolate from MT127116.1, MT159778.1, MT050414.1 and MT042777.1 showed the same genetic and mutation pathway with an evolutionary distance of 0.4 and 0.8 divergence time relative to the last common ancestor and the reference sequence.
Mutation and evolutionary pathway of the novel SARS-COV 2 coronaviruses: Figure 2 revealed that sequences retrieved from the NCBI gene bank for partial cdsRdRP region of SARS-COV 2 isolate from MT172668.1, MN938385.1 and LC522350.1 have the same genetic and mutation pathway with an evolutionary distance of 0.6 and 0.8 divergence time relative to the last common ancestor and reference sequence (Fig. 2). SARS-COV 2 sequences obtained from MT008022.1 isolate showed an evolutionary distance of 1.0 and a mutation pathway of 0.1 genetic divergence times relative to the last common ancestor. SARS-COV 2 sequences fromMT066157.1 and MN970003.1 isolates have a similar genetic and mutation pathway of 0.2 divergence time relative to the last common ancestor and with an evolutionary distance of 0.7 consensuses. The evolutionary distance of SARS-COV 2 sequences obtained from MT187977.1 isolate was 0.3 with a genetic and mutation pathway consensus of 0.1 relative to the last common ancestor (Fig. 2).
The study further revealed that the SARS-COV 2 S gene sequences of the isolates have a high and fast mutation rate from the reference sequences of SARS-COV 2 Beta viruses. This might explain why the viruses change in size, shape and structure thus making it difficult to produce a suitable vaccine from the reference sequence. This may also probably explain why the development of vaccines against most of the viral induce disease has not been possible due to the continuous metamorphosis and mutation of viruses in size, shape and protein structural coat32. This singular characteristic of the COVID 19 coronaviruses of the fast rate of mutation has made the production and development of vaccines (an attenuated form of the coronavirus) difficult as the mutant strains possess variable attenuation coefficients with which resurgence is possible after attenuation.
Genetic polymorphism/single nucleotide polymorphism (SNPs): Great genetic variability was observed among the mutant strains of the coronaviruses evaluated. This is polymorphism. The difference in single-nucleotide among the different isolates vertically in the same loci after multiple sequence alignment explains the variations in their characteristics. The SNPs are good molecular markers that can be employed to detect even the smallest variability among the coronaviruses strains. The genetic polymorphism and SNPs associated with coronaviruses as evident in this in silico study may affect drug response by influencing the action of the drug on the target molecule or the metabolism of the drugs on the coronavirus in vivo. Many SNPs have been found to correlate positively and strongly with drug inefficacy. Multiple nucleotide sequence alignments revealed that the SARS-COV 2 S gene sequences of the isolates have only three (3) conserve region at locus position 815 (Guanine conserve region), locus position 808 (Adenine conserve region) and locus position 679 (Guanine conserve region). The multiply aligned sequences showed more than 100 single nucleotide polymorphisms (SNPs) without gaps obtained from the coronaviruses evaluated thus indicating a high degree of genetic polymorphism among the novel pandemic coronaviruses. This characteristic of the coronaviruses explains its ability to overcome the antibody reactions of the host and other haptens like drugs. Multiple sequence alignment also revealed more nucleotide deletions which translate to the higher rate of mutation (Fig.2) observed among the isolates. The total number of mutation sites was 1218 with an average nucleotide diversity of 0.4221 for the coronavirus strains.
CONCLUSION
The study determined the genetic and thermal stability potentials of the isolates using the instability index, aliphatic index, guanine-cytosine contents, hydropathicity and half-life of the isolates in human reticulocytes cells in vitro. High ultraviolet light (extinction coefficient) exposures on any object harbouring the virus will breakdown the viral protein coat. The characteristics of the coronaviruses revealed their ability to change quickly producing many mutant copies of the coronavirus that are not exact, thus conferring on the mutant strains the ability to escape the host immune system. This thus calls for concerted efforts in studying the characteristics and mutation rates of the viruses to be able to predict the future mutation rate and attributes of the present strains to find a suitable therapy for the pandemic and biosecurity of humans against the virus in the future.
SIGNIFICANCE STATEMENT
This study discovered the genetic and thermal stability status of the coronaviruses and went ahead to validate some of the guidelines used as safety measures that can be beneficial for the general populace especially in the face of the global pandemic. This study will help the researcher to uncover the critical nature of the pathogens that many researchers were not able to explore. Thus unmasking the potential means for designing and developing prophylactic and therapeutic agents for the pandemic which the scientists are presently trying to arrive at.
REFERENCES
- Wu, F., S. Zhao, B. Yu, Y.M. Chen and W. Wang et al., 2020. A new coronavirus associated with human respiratory disease in China. Nature, 579: 265-269.
CrossRefDirect Link - Zhou, P., X.L. Yang, X.G. Wang, B. Hu and L. Zhang et al., 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579: 270-273.
CrossRefDirect Link - Holshue, M.L., C. DeBolt, S. Lindquist, K.H. Lofy and J. Wiesman et al., 2020. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med., 382: 929-936.
CrossRefDirect Link - Li, Q., X. Guan, P. Wu, X. Wang and L. Zhou et al., 2020. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med., 382: 1199-1207.
CrossRefPubMedDirect Link - Zhu, N., D. Zhang, W. Wang, X. Li and B. Yang et al., 2020. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med., 382: 727-733.
CrossRefDirect Link - Tamura, K., M. Nei and S. Kumar, 2004. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U.S.A., 101: 11030-11035.
CrossRefDirect Link - Tamura, K., G. Stecher, D. Peterson, A. Filipski and S. Kumar, 2013. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol., 30: 2725-2729.
CrossRefPubMedDirect Link - Chim, S.S.C., S.K.W. Tsui, K.C.A. Chan, T.C.C. Au and E.C.W. Hung et al., 2003. Genomic characterisation of the severe acute respiratory syndrome coronavirus of Amoy Gardens outbreak in Hong Kong. Lancet, 362: 1807-1808.
CrossRefDirect Link - Chen, N., M. Zhou, X. Dong, J. Qu and F. Gong et al., 2020. Epidemiological and clinical characteristics of 99 cases of 2019 novel Coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet, 395: 507-513.
CrossRefPubMedDirect Link - Huang, C., Y. Wang, X. Li, L. Ren and J. Zhao et al., 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet, 395: 497-506.
CrossRefDirect Link - Ziebuhr, J., V. Thiel and A.E. Gorbalenya, 2001. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J. Biol. Chem., 276: 33220-33232.
CrossRef - Chan, J.F.W., S. Yuan, K.H. Kok, K.K.W. To and H. Chu et al., 2020. A Familial Cluster of Pneumonia Associated with the 2019 Novel Coronavirus Indicating Person-to-Person Transmission: A Study of a Family Cluster. Lancet, 395: 514-523.
CrossRefDirect Link - Wölfel, R., V.M. Corman, W. Guggemos, M. Seilmaier and S. Zange et al., 2020. Virological assessment of hospitalized patients with COVID-2019. Nature, 581: 465-469.
CrossRefDirect Link - Yang, Z.Y., W.P. Kong, Y. Huang, A. Roberts, B.R. Murphy, K. Subbarao and G.J. Nabel, 2004. A DNA vaccine induces SARS coronavirus neutralization and protective immunity in mice. Nature, 428: 561-564.
CrossRefDirect Link - Yount, B., K.M. Curtis and R.S. Baric, 2000. Strategy for systematic assembly of large RNA and DNA genomes: Transmissible gastroenteritis virus model. J. Virol., 74: 10600-10611.
CrossRefDirect Link - Cho, K.O., A.E. Hoet, S.C. Loerch, T.E. Wittum and L.J. Saif, 2001. Evaluation of concurrent shedding of bovine coronavirus via the respiratory tract and enteric route in feedlot cattle. Am. J. Vet. Res., 62: 1436-1441.
CrossRefDirect Link - Scobey, T., B.L. Yount, A.C. Sims, E.F. Donaldson and S.S. Agnihothram et al., 2013. Reverse genetics with a full-length infectious cDNA of the middle East respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA, 110: 16157-16162.
CrossRefDirect Link - Yount, B., K.M. Curtis, E.A. Fritz, L.E. Hensley and P.B. Jahrling et al., 2003. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA, 100: 12995-13000.
CrossRefDirect Link - Chung, A.W., M.P. Kumar, K.B. Arnold, W.H. Yu and M.K. Schoen et al., 2015. Dissecting polyclonal vaccine-induced humoral immunity against HIV using systems serology. Cell, 163: 988-998.
CrossRefDirect Link - Deleage, C., A. Schuetz, W.G. Alvord, L. Johnston and X.P. Hao et al., 2016. Impact of early cART in the gut during acute HIV infection. JCI Insight, Vol. 1.
CrossRefDirect Link - Cho, K.O., M. Hasoksuz, P.R. Nielsen, K.O. Chang, S. Lathrop and L.J. Saif, 2001. Cross-protection studies between respiratory and calf diarrhea and winter dysentery coronavirus strains in calves and RT-PCR and nested PCR for their detection. Arch. Virol., 146: 2401-2419.
CrossRefDirect Link - Chouljenko, V.N., X.Q. Lin, J. Storz, K.G. Kousoulas and A.E. Gorbalenya, 2001. Comparison of genomic and predicted amino acid sequences of respiratory and enteric bovine coronaviruses isolated from the same animal with fatal shipping pneumonia. J. Gen. Virol., 82: 2927-2933.
CrossRefDirect Link - Zhong, N.S., B.J. Zheng, Y.M. Li, L.L.M. Poon and Z.H. Xie et al., 2003. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China. Lancet, 362: 1353-1358.
CrossRefDirect Link - Ziebuhr, J., E.J. Snijder and A.E. Gorbalenya, 2000. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol., 81: 853-879.
CrossRefDirect Link - Costantini, V., P. Lewis, J. Alsop, C. Templeton and L.J. Saif, 2004. Respiratory and fecal shedding of porcine respiratory coronavirus (PRCV) in sentinel weaned pigs and sequence of the partial S-gene of the PRCV isolates. Arch. Virol., 149: 957-974.
CrossRefDirect Link - de Haan, C.A.M., P.S. Masters, X. Shen, S. Weiss and P.J.M. Rottier, 2002. The group-specific murine coronavirus genes are not essential but their deletion, by reverse genetics, is attenuating in the natural host. Virology, 296: 177-189.
CrossRefDirect Link - Deleage, C., S.W. Wietgrefe, G.D. Prete, D.R. Morcock and X.P. Hao et al., 2016. Defining HIV and SIV reservoirs in lymphoid tissues. Pathog. Immun., 1: 68-106.
CrossRefDirect Link - Lin, J.R., B. Izar, S. Wang, C. Yapp and S. Mei et al., 2018. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife, Vol. 7.
CrossRefDirect Link - Tsang, K.W., P.L. Ho, G.C. Ooi, W.K. Yee and T. Wang et al., 2003. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med., 348: 1977-1985.
CrossRefDirect Link - Altfeld, M., T.M. Allen, X.G. Yu, M.N. Johnston and D. Agrawal et al., 2002. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature, 420: 434-439.
CrossRefDirect Link - Rockx, B., T. Kuiken, S. Herfst, T. Bestebroer and M.M. Lamers et al., 2020. Comparative pathogenesis of COVID-19, MERS and SARS in a nonhuman primate model. Science, 368: 1012-1015.
CrossRefDirect Link - Xiao, X., S. Chakraborti, A.S. Dimitrov, K. Gramatikoff and D.S. Dimitrov, 2003. The SARS-CoV S glycoprotein: Expression and functional characterization. Biochem. Biophys. Res. Commun., 312: 1159-1164.
CrossRefDirect Link