
Sentot Joko Raharjo
Academic of Pharmacy and Food Analysis, Putra Indonesia Malang, Jl. Barito No. 5 Malang, East Java, 65123, Indonesia
LiveDNA: 62.32620
Takheshi Kikuchi
Bioinformatic Laboratory, Graduate Life Science School, Ritsumeikan University, Biwako Kusatsu Campus, Shiga Prefecture, Japan
Fatchiyah Fatchiyah
Department of Biology, Faculty of Sciences, University of Brawijaya, Malang, East Java, Indonesia
Trends in Bioinformatics
Year: 2020 | Volume: 13 | Issue: 1 | Page No.: 18-26







ABSTRACT
Background and Objective: Alpha-bulnesene is one of the main components of patchouli oil, but only a few pieces of information are available that only prove its in vitro analysis. Its activity can be determined by in vitro/in silico analysis and their relationship. The aim of the study to understand the function of in silico and in vitro: Alpha-bulnesene (CID94275) from patchouli oil as prostaglandin H2 synthase (PGHS-1/PGHS-2) inhibitor selective, the element was assessed by means IC50, binding energy calculation and PGHS-1/PGHS-2 selectivity. Materials and Methods: The alpha-bulnesene fraction of patchouli oil was isolated using fractional vacuum distillation patchouli oil by PiloDist-104. The analysis of alpha-bulnesene fraction was done using GC-MS, IR spectrometry and NMR-spectrometry. The fraction was calculated by IC50 PGHS-1/PGHS-2 value through spectrophotometric test PGHS-ovine 760700. The binding energy of (with GBMV model solvent) of alpha-bulnesene with PGHS-1 and PGHS-2 was done using the molecular docking tools by Hex 8.0 and the interactions were further visualized by Discovery Studio Client 3.5 software. Results: The analysis of 8th-fractions proved the existence of the alpha-bulnesene. The fraction of alpha-bulnesene from patchouli oil was found at 82.12% (8th-fractions). IC50 of alpha-bulnesene fraction from PGHS-1 was found at 23.28μM and PGHS-2 was at 59.52 μM. The score of binding energy obtained from the calculation (with GBMV model solvent) of alpha-bulnesene showed result of PGHS-1 =-77.58 kcal Mol–1; PGHS-2 =-59.59 kcal Mol–1. Conclusion: An alpha-bulnesene fraction from patchouli oil has a ratio of IC50 and binding energy that was potentially used as PGHS-1 inhibitor selective.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Sentot Joko Raharjo, Takheshi Kikuchi and Fatchiyah Fatchiyah, 2020. In silico and in vitro: Alpha-bulnesene Fraction from Patchouli Oil as Prostaglandin H2 Synthase Inhibitor Selective. Trends in Bioinformatics, 13: 18-26.
DOI: 10.3923/tb.2020.18.26
URL: https://scialert.net/abstract/?doi=tb.2020.18.26
DOI: 10.3923/tb.2020.18.26
URL: https://scialert.net/abstract/?doi=tb.2020.18.26
INTRODUCTION
Alpha-bulnesene is one of the major components of patchouli oil sesquiterpenoid groups from Pogostemon cablin Benth1. This compound has the bioactivity ability as an anti-aggregation platelet on rabbit blood through PAF (platelet-activating factor) inhibitor mechanism and PGHS enzyme2. Some of the available information only proves the in vitro analysis of activity of alpha-bulnesene. Furthermore, the activity of this compound can be studied through an analysis involving the in vitro and in silico which can demonstrate the relationship between the two methods of analysis.
Drugs that inhibit the mechanism of isoenzymes PGHS (prostaglandin H2 synthase) are called NSAID (non-steroid anti-inflammatory drugs). The prostaglandin enzyme of the H2 synthase pathway (or COX (cyclooxygenase) pathway) is the enzyme that is responsible for conducting the steps in the prostaglandin biosynthesis process, generating the product of prostaglandin-H2 (PGH2)3. There are two isoforms of PGHS enzymes namely: PGHS-1 and PGHS-24. PGHS-1 catalyzes the formation of prostaglandins to implement the regulation of physiological functions, meanwhile, PGHS-2 catalyzes the formation of "bad prostaglandins" which causes inflammation5. Inhibition of PGHS-2 produces analgesic, antipyretic and anti-inflammatory (NSAID). Whereas, the inhibition of PGHS-1 is responsible for providing the antithrombotic effect. Acetosal is one of NSAID medicines that is used as an anti-aggregation platelet which prevents the occurrence of platelet aggregation that can cause blockages in the blood vessels6-9. However, some classical NSAID drugs cause toxicity in the gastrointestinal tract, such as acetylsalicylic acid, phenylbutazone, indomethacin, diclofenac, ibuprofen and naproxen10,11. These drugs are non-selective for PGHS-1 and PGHS-2. The strategy undertaken to reduce the toxicity of classic NSAIDs can be done by developing the drugs as a selective inhibitor of PGHS-212,13. Steroids have an obvious role in the treatment of inflammatory diseases, but due to their toxicity, they can only be used within short periods except for very serious cases in which risks are acceptable. Consequently, the side effects of this drug cause great concern. Therefore, it is necessary to develop other methods or compounds made of natural ingredients that function as drugs inhibiting the PGHS-1/PGHS-2 selective.
The determination in vitro analysis of PGHS inhibitor selective used the IC50 (inhibitor concentration-50) and Selectivity Index (SI). Determination of IC50 analysis performed in vitro and in vivo used multiple detection methods PGHS activity, such as oxygen absorption method, peroxide method, an enzyme immunoassay (EIA) and radioimmunological assay (RIA)7[7]. The selectivity of cyclooxygenase can be supported by virtual modeling. The development of virtual modeling methods screens the docking results of drug compounds (ligands) of receptor protein to predict the position and the orientation (pose) of ligand interaction toward the target protein that has low molecular weight. This is a basic guideline to obtain the structure-activity relationship in case the condition of the high-resolution structure of a compound cannot be obtained. The development of virtual modeling is conducted to perform energy calculations for the complexes, protein and ligand using certain solvent models14-16. Bioactivities of some compounds isolated from natural products, organic materials, such as alpha-bulnesene isolated from patchouli oils can be used for inhibiting the PGHS-1/PGHS-2 isoenzyme unclearly.
The aim of this study was to qualitatively calculate the IC50 and binding energy for the selectivity of alpha-bulnesene (CID94275) inhibits prostaglandin H2 synthase (PGHS/COX). This research is expected to explore the selectivity of alpha-bulnesene as the inhibitor PGHS-1 and PGHS-2 through the IC50 determination and binding energy. Besides, this research also attempts at employing the approaching methods to determine the bioactivity of compounds that can be used as inhibitors made of natural organic materials.
MATERIALS AND METHODS
Study area: The study was carried out at the Academic of Pharmacy and Food Analysis of Putra Indonesia Malang, East Java and Chemical Research Center, LIPI, Serpong, West Java, Indonesia from March, 2019 to April, 2020.
Alpha-bulnesene fraction analysis: Patchouli oils were obtained from the steam distillation of Pogostemon cablin Benth in the home industry Trenggalek, Indonesia. The fraction of alpha-bulnesene was used in the fractional vacuum distillation using PiloDist-104-Germany. The operating conditions of a fractional-vacuum distillation performed in this research were determined based on the Identification of Compounds of patchouli oil. The operating conditions were set at pressure 1 mBar; The head temperature fractions (1st fraction to 8th fraction) were 95.0-122.0, 112.0-120.3, 120.3-111.7, 111.7-114.8, 114.8-112.3, 112.3-116.2, 116.2-118.7 and 118.7-124.6, respectively and the Flash Temperature fractions were set at 148.9-136, 156.0-154.0, 154.0-153.0, 153.0-162.9, 155.0-160.0, 160.0-161.0, 161,0-165.2 and 165.2-180.0, respectively. The fraction was analyzed through the GC-MS Shimadzu QP-2010 SE (Shimadzu Corporation Japan) with RTX-Wax column (Restek. Germany), MS detector1 and Wiley8 Library, NMR spectrometer JEOL Delta2 500 Hz (JEOL Ltd, Japan) and FT-IR spectrometer Shimadzu-8400 (Shimadzu Corporation Japan).
In silico analysis: Binding energy (Ebinds) PGHS-1 and PGHS-2 of alpha-bulnesene: 3D model from PDB ID: 1PTH was obtained from the SWISS-MODEL repository for PGHS-1 and 3D model from PDB ID: 6COX for PGHS-2. Alpha-bulnesene (CID94275) was downloaded from http://pubchem.ncbi. nlm.nih.gov/in a 3D-SDF format in which energy form was then minimized and converted to 3D-PDB format by Open Babel 2.3.1 in Hex 8.0 as the ligand for virtual screening. Hex8.0 Cuda software was used to calculate the docking ligand single alpha-bulnesene (CID94275) to PGHS-1 and PGHS-2. The output of the docking analyzed the 2D interaction of active site using Discovery Studio 2019 Client software to simulate all complex structures CID94275 to interact PGHS-1 and PGHS-2 with MM-GBMV model solvent. Furthermore, energy analysis (complex, protein and ligand) as a candidate model of PGHS-1 and PGHS-2 inhibitor selective was done using the Discovery Studio 2019 Client software. Binding energy was calculated based on the Eq.17,18:
ΔG = Gcomplex - [Gprotein + Gligand]
In vitro analysis: IC50 PGHS-1 and PGHS-2 of alpha-bulnesene fraction: The determination IC50 alpha-bulnesene fraction was done using the test of spectrophotometric COX (ovine). The concentration inhibitor, i.e.: 20, 60 and 180 μM in DMSO solvent. Pre-test preparation, protocol and analysis of the determination of IC50 were done through manual tests of spectrophotometric PGHS (ovine) Inhibitor Screening Assay Kit Series 760700.
Selectivity alpha-bulnesene as PGHS-1 and PGHS-2 inhibitor: The selectivity of alpha-bulnesene to PGHS-1/PGHS-2 inhibitor can be determined based on this equation: log ratio of (IC50 PGHS-2 and IC50 PGHS-1) and-log [ratio binding energy PGHS-2 and binding energy PGHS-1]7.
RESULTS
Alpha-bulnesene fraction analysis: The fraction of alpha-bulnesene (8th fraction) and the other fraction from patchouli oil was used in the vacuum fractional distillation using PiloDist-104, Germany. The analyses of alpha-bulnesene fraction, patchouli oil and the other fraction were done using GC-MS, which result is presented in Fig. 1a. The fragmentation analysis of alpha-bulnesene (CID94275) using GC-MS, is shown in Fig. 1b-1. The results of the mass spectrometry analysis of the alpha-bulnesene using GC-MS showed TIC retention time = 23.25 min. EI mass spectrum analysis showed results of m/z: 204 (base), 189, 175, 161, 147, 133, 119, 105, 93, 79, 67, 55. The simulation of the fragmentation pattern is also presented in Fig. 1b-1. The fragmentation pattern of this compound was simulated as follows: Start with the termination of these compounds through the release of CH3 radicals in the peak molecular ion (M+), to make the peak at m/z = (M+-15) = 189. The peak of m/z = M+-15 = 189, The-CH2 release occurred successively to obtain peak m/z = 133 which was followed by the release of-C2H4 to obtain the peak of m/z = 107. The peak m/z = 147 was brought to experience the release of-C2H8 to obtain peak m/z = 119, followed by the release of C2H2 to obtain peak m/z = 91, as shown in Fig. 1b-1. UV-Vis Spectra: no-absorption (structure of alpha-bulnesene does not have auxochromes and chromophore groups). IR Spectra (neat) υmax (KBr) cm–1 = 3072, 2991, 2801, 2720, 1776, 1613, 1442, 1377, 1212 was indicated by alkenyl CH stretch, alkenyl C=C stretch, alkyl, C-H stretch and fingerprint area. The analysis of NMR spectrum was exhibited in two broad singlet’s at δ/ppm 2.32-2.18 (2H, exocyclic methylene) and two near singlet’s at δ/ppm 0.90 and 0.84 (6H, two methyl groups attached to double bonds), as shown in Fig. 2b. Spectra data of EIMS, 1H-NMR and 13C-NMR were fairly in line with the results of elucidation structure results reported in the literature (as alpha-bulnesene) as shown in Fig. 1b-2.
In vitro analysis: IC50 of alpha-bulnesene to PGHS-1 and PGHS-2: The values IC50 of 8th fraction (alpha-bulnesene fraction) were calculated using the test kit PGHS-Ovine. The regression linear analysis was done to determine the IC50 values of alpha-bulnesene fraction to PGHS-1 and PGHS-2, as shown in Fig. 2a.
In silico analysis: Binding energy (Ebinds) of alpha-bulnesene to PGHS-1 and PGHS-2: Docking (ligands to proteins) alpha-bulnesene (CID94275) to PGHS-1 and PGHS-2 was done using Hex 8.0 software and visualization using Discovery Studio 2019 Client software, as shown in Fig. 2b. The software of Discovery Studio 2019 Client was also used to simulate the complexity of ligand alpha-bulnesene interacts with proteins PGHS-1 and PGHS-2 with MM-GBMV model solvent. Binding energy calculations using the Eq:
ΔG = Gcomplex - [G-protein + Gligand]
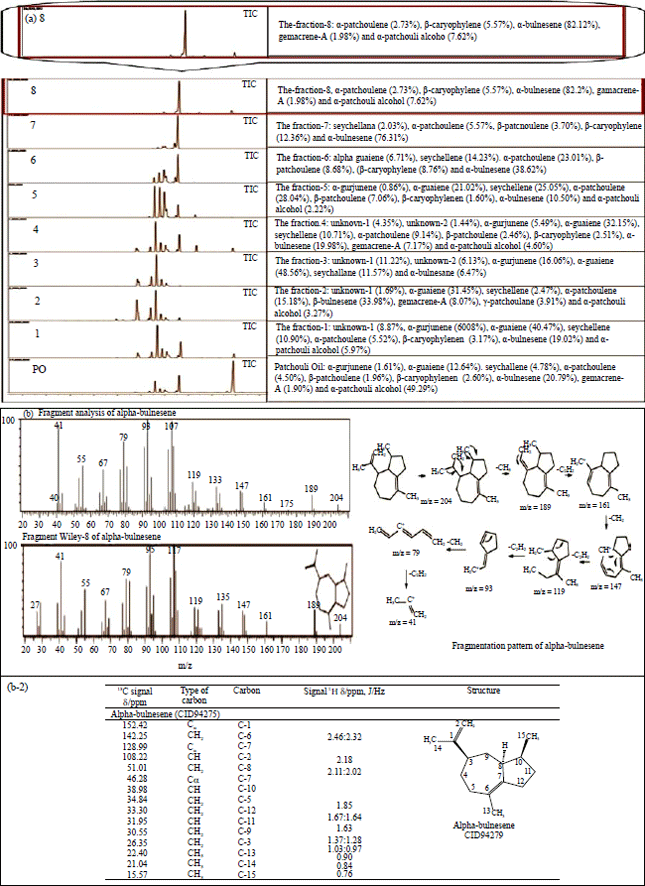 | |
| Fig. 1(a-b): | Analysis of alpha-bulnesene (8th) fraction |
(a) TIC fraction-8 (alpha-bulnesene), patchouli oil and the other fraction analyzed by GC-MS Shimadzu QP-2010 with RTX-Wax column, detector MS and Wiley8 Library, (b) MS and NMR alpha-bulnesene analyses, (b-1) The fragmentation alpha-bulnesene and the fragmentation pattern of alpha-bulnesene, (b-2) 1H-NMR and 13C-NMR alpha-bulnesene analyses | |
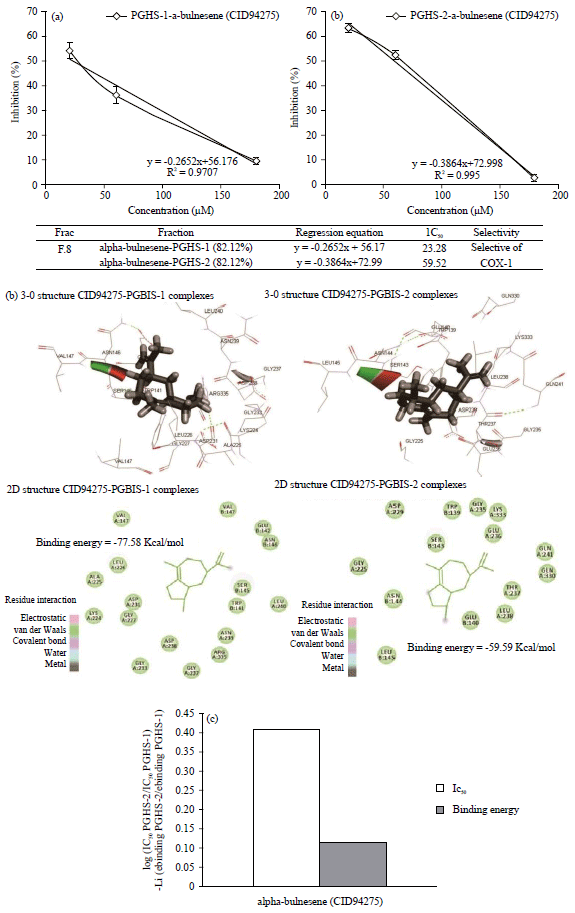 | |
| Fig. 2(a-c) | In silico and in vitro analyses of alpha-bulnesene (CID94275) to PGHS-1/PHGS-2 |
(a) In silico : 2D and 3D-structure ligand alpha-bulnesene interaction with PGHS-1 and PGHS-2, (b) Regression analysis of IC50 of alpha-bulnesene fraction-PGHS complexes and (c) Selectivity of IC50 and binding energy alpha-bulnesene (CID94275) to PGHS-1/PGHS-2 | |
The score of binding energy calculation (with GBMV model solvent) of alpha-bulnesene from PGHS-1 was found at -77.58 kcal Mol–1 and from PGHS-2 at-59.59 kcal Mol–1, as shown Fig. 2b.
Selectivity alpha-bulnesene as PGHS-1 and PGHS-2 inhibitor: According to the equation-log ratio [IC50 PGHS-2 and IC50 PGHS-1] and-log [ratio binding energy PGHS-2 and binding energy PGHS-1], a selectivity of alpha-bulnesene to PGHS-1/PGHS-2 inhibitor is presented in Fig. 2c.
DISCUSSION
Patchouli oil (volatile compounds) consists of rich sesquiterpenoid, mainly the patchouli alcohol (patchoulol) and some tricyclic sesquiterpenoid, such as alpha-bulnesene, alpha-guaiene and seychellene. The other components include alpha-patchoulene, beta-patchoulene, beta-caryophyllene, germacrene-A, alpha-gurjunene and the sesquiterpenoids1. ISO Standard 3757:2002 patchouli oil, the major constituent of patchouli oil were (alpha-patchouli alcohol 30-35%), alpha-bulnesene (13-21%) and alpha-guaiene (11-16%)17-19. Tsai, et al.2, has analyzed the compounds of patchouli oil (Pogostemon cablin Bentham (Lamiaceae)) form Koda production Pharmaceutical Co., Ltd. Taiwan which consisted of alpha-guaiene (20.62%), alpha-bulnesene (16.18%), beta-patchoulene (12.12%) and patchouli alcohol (11.12%). In this study, GC-MS analysis of patchouli oil compounds from traditional steam distillation indicated an increase in the content of compounds, such as alpha-patchouli alcohol, alpha-bulnesene, alpha-guaiene and seychellene, respectively, as shown in Fig. 1a. The characteristics of patchouli oil compounds obtained from Trenggalek-East Java have major components, including alpha-patchouli alcohol, alpha-bulnesene, alpha-guaiene and seychellene, respectively. The patchouli oil contains the compounds that conform to Standard ISO patchouli oil1,20-24.
The properties of alpha-bulnesene, C15H24; weight = 204.351 [g Mol–1]; XlogP = 3.74; Gibbs energy = 77.7 [kcal Mol–1], complexity 295.16, H-donor = 0 and H-receptor = 021,22. In this research, the isolation alpha-bulnesene from patchouli oil by fractional-vacuum distillation (PiloDist-104) has obtained fraction -8 to 82.12%. The presence of alpha-bulnesene in the 8th fraction was seen from the result of GC-MS analyzes. Data EI-MS of fraction-8 for alpha-bulnesene as a member of sesquiterpenoid showed a decreasing peak similar to Wiley8 Library. According to Silverstein et al.25, the decreasing peak m/z = 14 indicates that the compound belongs to the saturated hydrocarbon group, including the sesquiterpenoid group. The results of mass spectra, IR spectra and NMR analysis show similar results to the results of previous research which states that fraction-8, a yellow liquid and exhibited the expected spectral properties: MS, m/Z 316 (M+ = 75), 180 (60), 167 (100), 150 (37), 137 (71); IR Spectra (neat) υmax (KBr) cm–1 = 3072 (=C-H stretch medium), 2991 (-C-H stretch weak), 2801 (C-H strong), 2720 (C-H stretch alkane), 1776 (cyclic stretch), 1613 (C=C stretching cyclic alkene), 1442 (C-H bending alkane), 1377 (C-H bending alkane) and 1212 (C-H bending alkane); NMR spectrum, two broad singlet’s at τ 5.36-5.62 (2H, exocyclic methylene) and two near-singlet’s at τ 8.37 (6H, two methyl groups attached to double bonds), which strongly support the structure of alpha-bulnesene19. In this research, it is found that the characteristic of alpha-bulnesene fraction (8th fraction) from patchouli oil (from Pogostemon cablin Benth (East Java-Indonesia)) obtained major components of alpha-bulnesene20-24 and some other components such as impurities, as shown in Fig. 1. The Hsu research, the isolated alpha-bulnesene obtained sub-fraction-3.5 from fraction-3 (fraction -1 to 4) by GC-TCD as performed by a Hewlett-Packard 5890 gas chromatography equipped with a thermal conductivity detector. The oven temperature was set at 50°C for 4 min, then it was programmed from 50-272°C, at 6°C/min and finally set at 272°C for 3 min19. Other studies showed that the alpha bulnesene fraction was separated using chromatographic techniques, while our study employed fractional distillation, where the yield of alpha-bulnesene fraction preponderant1,24.
Non-steroidal anti-inflammatory drugs (NSAIDs) have a huge therapeutic benefit for treatments of various types of inflammatory conditions. The target for these drugs is the PGHS, a rate-limited enzyme involved in the conversion of arachidonic acid into inflammatory prostaglandins. PGHS-2 selective inhibitors are believed to have the same anti-inflammatory, anti-pyretic and analgesic effects such as those of non-selective inhibitor NSAIDs with little or none of the gastrointestinal side effects. The PGHS-2 and PGHS-1 selectivity ratios are vital in the design of PGHS-2 inhibitory drugs. Natural product-based compounds seem to be better as these compounds are generally supposed to devoid severe side effects23. The PGHS-2 and PGHS-1 selectivity ratio determine the ratio of IC50 PGHS-2/PGHS-1. One method to determine the IC50 values is using the peroxidase method. This assay measures the peroxidase catalytic activity of PGHS as an indirect measurement of PGHS activity that will be very sensitive to compounds that are also co-reductions. Meanwhile, the cyclooxygenase initiation is dependent on peroxidase turnover, the inverse is not true: Peroxidase activity can occur at/or near-maximal efficiency within the absence of a functional cyclooxygenase active site. It should be also noted that the most precise enzymatic measurements made in the literature are derived from this assay, which directly quantitates the two activities of the enzyme7. This research, the 8th fraction (fraction of alpha-bulnesene) have the value IC50 of PGHS-1 (23.28 μM) and PGHS-2 (59.52 μM), therefore the selectivity of the alpha-bulnesene fraction is inclined as PGHS-1 inhibitors. Some literature also indicates that the compound is generally natural-based products consisting of mainly PGHS-1. This result goes consistently with the result of Tsai et al.2, research which states that alpha-bulnesene inhibited AA-induced thromboxane B(2) (TXB(2)) formation and prostaglandin E(2) (PGE(2)) formation. These results indicate that the inhibitory effect of alpha-bulnesene on platelet aggregation occurred due to the dual activity; specifically, the chemical blocked PAF-induced intracellular signal transduction and interfered with cyclooxygenase activity, which resulted in the decrease of thromboxane formation. This study is the first study that demonstrates alpha-bulnesene as a PAF receptor antagonist as well as an anti-platelet aggregation agent while Park et al.20, have examined the in vitro, ex vitro and in vivo use of the herbal medicine (including P. cablin) as a novel antithrombotic agent. Thus, our research supports the mechanism of the inhibition of PGHS-1. The anti-platelet activity has been known to occur due to the induction of intracellular signal transduction by alpha-bulnesene which interferes with the activities of cyclooxygenase which result in the decrease of thromboxane production. The IC50 values in the PGHS need conformation in silico analyses.
The result of in silico analysis supported the conformational ability analysis of alpha-bulnesene as the candidate of inhibitor selective. The further confirmation in this study was conducted by docking (ligands to proteins) ligand alpha-bulnesene (CID94275) to PGHS-1 and PGHS-2, visualization analysis and calculations of binding energy. The visualizations of active site interactions between alpha-bulnesene-PGHS-1 and-PGHS-2 complexes showed all van Der Walls interaction because alpha-bulnesene does not have any H-donor and H-acceptor20-23. The different types of interactions in the alpha-bulnesene-PGHS-1 and alpha-bulnesene-PGHS-2 complexes will certainly affect its binding free energy23. Binding energy was performed using the MM-GBMV model solvent method. MM-Generalized Born Molecular Volume (GBMV) models provide a computationally efficient means to represent the electrostatic effects of solvent and are widely used. A class of particularly fast GBMV models is based on the integration of an interior volume which is approximated as a pairwise union of atom spheres-effectively, in which the interior is defined by a van der Waals interaction rather than Lee-Richards molecular surface. The approximation is computationally efficient, but if it is uncorrected, it will allow high dielectric (water) regions to become smaller than the water molecule among atoms, leading to the decreased accuracy. An earlier pairwise of the GBMV model was developed using a simple analytic correction term that largely alleviates the problem by correctly describing the solvent excluded volume of each pair of atoms. The correction term introduces a free energy barrier to the separation of non-bonded atoms. This free energy barrier is seen in explicit solvent and Lee-Richards molecular surface implicit solvent calculations. The robustness and simplicity of the correction preserve the efficiency of the pairwise GBMV models while making them a better approximation to reality26. The calculation of binding free energy is computed as follows:
G = Gcomplex - [G-protein + Gligand]
where, Gcomplex is the absolute free energy of the complex, Gprotein is the absolute free energy of the protein and Gligand is the absolute free energy of the ligand)20-23. The free energy of each term was estimated as a sum of the three terms:
[G] = [EMM] + [GSOL] - T.[S]
where, EMM is the molecular mechanic’s energy of the molecule expressed as the sum of the internal energy (bonds, angles and dihedrals) (Eint), electrostatic energy (Eele) and van der Waals term (Evdw):
[EMM] = [Eint] + [Eele] + [Evdw]
〈GSOL〉 accounts for the solvation energy which can be divided into the polar and nonpolar part. The polar part accounts for the electrostatic contribution to solvation and is obtained by solving the linear Poisson Boltzmann equation in a continuum model of the solvent. On the other hand, the other part accounts for the nonpolar contribution to solvation and represents the cost of creating a cavity inside the solvent. This is linearly related to the solvent accessible surface area. [GSOL] implicitly includes the entropy, unlike 〈EMM〉. Finally, configurationally entropies were computed by diagonalization of the cartesian coordinate covariance matrix following the method described by Schlitter and extensively tested in protein systems. Scoring binding energy calculation (with GBMV model solvent) of alpha-bulnesene (PGHS-1 = -77.58 kcal Mol–1; PGHS-2 = -59.59 kcal Mol–1). The value similar with the selective IC50 value for alpha-bulnesene, as shown in Fig. 2c. Therefore, alpha-bulnesene has been indeed proven to act as a selective inhibitor of PGHS-1. The similar research to the natural product as PGHS inhibitor selective showed the binding energy calculation of some alpha-patchouli alcohol isomer which was also suggested as candidates for a PGHS-1 inhibitor selective27. The relationship binding energy, Ki and IC50 are defined Gbinding = 2,303 R.T log Ki.
This study only shows the relationship between binding energy and IC50 value. However, both relationship does not present the interactions of the substrate and the inhibitors to act as a competitive inhibitor, un-competitive and non-competitive against the enzyme. This study can be expanded further by doing conformation again to the relationship of the IC50 value and the binding energy. Alpha-bulnesene is one result of patchouli oil from Pogostemon cablin Benth. This compound is a natural product compound. This compound has been proven to be also one of the compounds has the ability as PGHS-1 selective. Through semi-synthetic small changes in their structure, their selectivity against PGHS-2 can be improved. This study illustrates one way of the characterization of potential natural PGHS inhibitors.
CONCLUSION
In silico/in vitro analysis: Alpha-bulnesene (CID94275) from patchouli oil as prostaglandin H2 synthase (PGHS-1/PGHS-2) has IC50 values from PGHS-1 was found at 23.28 μM and PGHS-2 was at 59.52 μM, while the score of binding energy obtained from the calculation (with GBMV solvent model) of alpha-bulnesene showed the result of PGHS-1 = -77.58 kcal Mol–1; PGHS-2 = -59.59 kcal Mol–1, so alpha-bulnesene fraction from patchouli oil that was a candidate as a PGHS-1 selective inhibitor.
SIGNIFICANCE STATEMENT
This research has the advantage of having examined the ability alpha-bulnesene isolate from the patchouli oil that determines their nature as PGHS-1 protein inhibitors. In vitro analysis, alpha-bulnesene isolates showed their ability as PGHS-1 inhibitors. This is also synergistically supported in its in silico analysis. This study will help researchers to uncover the role of natural material compounds that many researchers cannot yet explore. Thus, this computational approach can emphasize researchers around the world designing anti-inflammatory drugs based on active compounds of natural ingredients from sources that are not/under-exploited. However, although it is effective in the in vitro and in silico analysis of this study, further in need of an in vivo investigation as a candidate for effective anti-inflammatory drugs.
ACKNOWLEDGMENTS
The authors acknowledge all facilities of Bioinformatics Laboratory Department of Creation Core, Ritsumeikan University, for providing the in silico analyses. Special thanks are due to Masanari Matsuoka, Antonius Christianto, Michirou Kabata, Sayaka Ohara, Yousuke Kawai and working group Bioinformatics Laboratory, Biwako Kutsasu Campus, Ritsumeikan University, for helpful support analysis and discussions.
REFERENCES
- Pawestri, A.M. and N. Fitri, 2019. Effect of Aspergillus niger on fermentation process in increasing the quality of patchouli oil. EKSAKTA: J. Sci. Data Anal., 19: 15-25.
CrossRefDirect Link - Tsai, Y.C., H.C. Hsu, W.C. Yang, W.J. Tsai, C.C. Chen and T. Watanabe, 2006. α-bulnesene, a PAF inhibitor isolated from the essential oil of Pogostemon cablin. Fitoterapia, 78: 7-11.
CrossRefDirect Link - Demirayak, S., R. Beis, A.C. Karaburun, I. Kayagil, Z. Incesu and U. Ucucu, 2006. Synthesis and cytotoxic and analgesic activities of some 1, 5-diaryl-3-ethoxycarbonylpyrrole derivatives. J. Enzyme Inhib. Med. Chem., 21: 113-118.
CrossRefDirect Link - Antman, E.M., D. DeMets and J. Loscalzo, 2005. Cyclooxygenase inhibition and cardiovascular risk. Circulation, 112: 759-770.
CrossRefDirect Link - Perrone, M.G., A. Scilimati, L. Simone and P. Vitale, 2010. Selective COX-1 Inhibition: A therapeutic target to be reconsidered. Curr. Med. Chem., 17: 3769-3805.
CrossRefDirect Link - Blobaum, A.L. and L.J. Marnett, 2007. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem., 50: 1425-1441.
CrossRefPubMedDirect Link - Moth, C.W., J.J. Prusakiewicz, L.J. Marnett and T.P. Lybrand, 2005. Stereoselective binding of indomethacin ethanolamide derivatives to cyclooxygenase-1. J. Med. Chem., 48: 3613-3620.
CrossRefDirect Link - Dubois, R.N., S.B. Abramson, L. Crofford, R.A. Gupta, L.S. Simon, L.B.V.D. Putte and P.E. Lipsky, 1998. Cyclooxygenase in biology and disease. FASEB J., 12: 1063-1073.
CrossRefPubMedDirect Link - Kartasasmita, R.E., R. Herowati, N. Harmastuti and T. Gusdinar, 2009. Quercetin derivates docking based on study of flavanoids interaction to cyclooxygenase-2. Indones. J. Chem., 9: 297-302.
CrossRefDirect Link - Alberto, M.R., I.C. Zampini and M.I. Isla, 2009. Inhibition of cyclooxygenase activity by standardized hydroalcoholic extracts of four Asteraceae species from the Argentine Puna. Brazilian J. Med. Biol. Res., 42: 787-790.
CrossRefDirect Link - Limongelli, V., M. Bonomi, L. Marinelli, F.L. Gervasio, A. Cavalli, E. Novellino and M. Parrinello, 2010. Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. PNAS, 107: 5411-5416.
CrossRefDirect Link - Haider, M.K., H.O. Bertrand and R.E. Hubbard, 2011. Predicting fragment binding poses using a combined MCSS MM-GBSA approach. J. Chem. Inf. Model., 51: 1092-1105.
CrossRefDirect Link - Campanera, J.M. and R. Pouplana, 2010. MMPBSA decomposition of the binding energy throughout a molecular dynamics simulation of amyloid-beta (Aβ10-35) aggregation. Molecules, 15: 2730-2748.
CrossRefDirect Link - Uciechowska, U., J. Schemies, M. Scharfe, M. Lawson, K. Wichapong, M. Jung and W. Sippl, 2012. Binding free energy calculations and biological testing of novel thiobarbiturates as inhibitors of the human NAD+ dependent histone deacetylase Sirt2. Med. Chem. Commun., 3: 167-173.
CrossRefDirect Link - Chen, Y., Y. Wu, Y. Xu, J.F. Zhang, X.Q. Song, G.P. Zhu and X.W. Hu, 2014. Dynamic accumulation of sesquiterpenes in essential oil of Pogostemon cablin. Rev. Bras. Farmacogn., 24: 626-634.
CrossRefDirect Link - Tsai, Y.C., H.C. Hsu, W.C. Yang, W.J. Tsai, C.C. Chen and T. Watanabe, 2006. α-bulnesene, a PAF inhibitor isolated from the essential oil of Pogostemon cablin. Fitoterapia, 78: 7-11.
CrossRefDirect Link - Raharjo, S.J. and F. Fatchiyah, 2013. Virtual screening of compounds from the patchouli oil of Pogostemon herba for COX-1 inhibition. Bioinformation, 9: 321-324.
CrossRefDirect Link - Raharjo, S.J., C. Mahdi, N. Nurdiana, T. Kikuchi and F. Fatchiyah, 2017. In vitro and in-silico: Selectivities of seychellene compound as candidate cyclooxygenase isoenzyme inhibitor on pre-osteoblast cells. Curr. Enzyme Inhib., 13: 2-10.
CrossRefDirect Link - Raharjo, S.J., C. Mahdi, N. Nurdiana, T. Kikuchi and F. Fatchiyah, 2014. Binding energy calculation of patchouli alcohol isomer cyclooxygenase complexes suggested as COX-1/COX-2 selective inhibitor. Adv. Bioinform.
CrossRefDirect Link - Raharjo, S.J., 2019. Virtual Screening of Sesquiterpenoid Pogostemon herba as Predicted Cyclooxygenase Inhibitor. In: Molecular Docking and Molecular Dynamics. Stefaniu, A., (Ed.)., IntechOpen, UK.
CrossRefDirect Link - Swamy, M.K. and U.R. Sinniah, 2015. A comprehensive review on the phytochemical constituents and pharmacological activities of Pogostemon cablin Benth.: An aromatic medicinal plant of industrial importance. Molecules, 20: 8521-8547.
CrossRefDirect Link - Mongan, J., C. Simmerling, J.A. McCammon, D.A. Case and A. Onufriev, 2010. Generalized born model with simple, tobust molecular volume correction. J. Chem. Theory Comput., 3: 156-169.
CrossRefDirect Link - Cer, R.Z., U. Mudunuri, R. Stephens and F.J. Lebeda, 2014. IC50-to-Ki: A web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res., 37: 441-445.
CrossRefDirect Link