
Bhaludra Chandra Sekhar Singh
Department of Plant Science, College of Agriculture and Veterinary Sciences, Ambo University, Post Box: 19, Ambo, Ethiopia, East Africa
LiveDNA: 91.10338
Trends in Bioinformatics
Year: 2020 | Volume: 13 | Issue: 1 | Page No.: 10-17







ABSTRACT
Background and Objective: Homology modeling assumes a focal job in deciding protein structure in the basic genomics venture refers to modeling of the 3D structure of a protein by exploiting structural information from the known configurations of similar proteins. The docking studies are the inclusion of receptor flexibility is an important role because of the major consequences on molecular recognition binding sites of many therapeutic targets sample, which has a wide range of conformational states to make use of very large sets of X-ray structures and to assess the execution of flexible receptor docking in binding-mode prediction virtual screening experiments. To analyze the homology modeling of a protein 3D structure, this is helpful to design a drug. Materials and Methods: The homology model is evaluated based on a template for identification, alignments for single or multiple sequences, target 3D structure, model refinement and model validation. The on-going advances in homology modeling that improve the accuracy of modeling results especially in recognizing and adjusting successions to layout structures, far off homologs, modeling of loops and side chains just as identifying mistakes in a model added to the reliable forecast of protein structure. Results: A model is said to be legitimate just when a couple of mutilations in nuclear contacts are available. The Ramachandran plot is presumably the most dominant determinant of the nature of the protein. Conclusion: Protein structure prediction has been a fantasy for an academic network for decades, for computational scientific experts, yet also for physicist, mathematician and PC researcher.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Bhaludra Chandra Sekhar Singh, 2020. Study on Role of Homology in Prediction of Protein Structure and Challenges of Homology. Trends in Bioinformatics, 13: 10-17.
DOI: 10.3923/tb.2020.10.17
URL: https://scialert.net/abstract/?doi=tb.2020.10.17
DOI: 10.3923/tb.2020.10.17
URL: https://scialert.net/abstract/?doi=tb.2020.10.17
INTRODUCTION
Homology modeling has turned into a helpful apparatus for the expectation of protein structure when just grouping information is accessible. Homology modeling is conceivably a valuable apparatus for the mycologist, as the quantity of fungal gene groupings accessible has detonated as of late, while the quantity of tentatively decided fungal protein structure stays low. Projects accessible for homology modeling use various methodologies and techniques to create the last model1. The undeniable favorable position is that the homology modeling method loosens up the stringent necessity of force field and gigantic confirmation looking since it abstains from the computation of a physical chemistry force field and replaces it, in huge part, with the tallying of succession personalities2. Auxiliary information is frequently more significant than arrangement alone for deciding protein work. The sequence identity and the binding site area is more than 50% of active compounds between the template and model3. The overall Protein Data Bank (wwPDB consortium 2019) contains roughly 144,000 tentatively decided protein three dimensional (3D) structures and small molecule bounds with nucleic acids or protein4. This developing hole between the arrangements accessible and the protein 3D structures decided is in a disturbing condition5. For decades, structure forecast has entranced mainstream researchers; it is a critical issue that is easy to characterize however hard to tackle. The reliable testing of homology-based algorithms is based on 3D structures of protein-protein complexes. The results of 3D structures protein complexes known as Homology-Based Complex Prediction models compared to the generated were models6. The present challenges in docking studies are the inclusion of receptor flexibility is the pivotal role because the major consequences on molecular recognition binding sites of many therapeutic targets sample, which has a wide range of conformational states to make use of very large sets of X-ray structures of heat shock protein 90 (HSP90) and cyclin-dependent kinase 2 (CDK2) to assess the execution of flexible receptor docking in binding-mode prediction virtual screening experiments and flexible receptor docking performs remedial than rigid receptor docking in the earlier application7. In actuality, homology modeling has accepted an inexorably significant job in protein structure forecast in the ongoing years with the approach of basic genomics activities around the globe. This transformative relationship gives the basis to basic genomics, an efficient and enormous scale effort towards the basic portrayal of all things considered, where a delegate protein in every family is picked to be settled tentatively with the rest dependably anticipated by a homology modeling technique8. At present, just 7677 protein families have been distinguished by the Pfam database. Unquestionably, this number is unequivocally subject to the arrangement likeness shorts used to group the succession space. Sequence-structure deficit marks one of the critical problems in the current situation where high-efficiency sequencing has resulted in large datasets of protein sequences, but their corresponding 3D structures still need to be resolved9. Homology modeling was found to give 3D structures with the most astounding exactness. Besides, it is a protein 3D structure expectation technique that requirements less time and lower cost with clear advances. In this manner, homology modeling is generally utilized for the generation of 3D structures of proteins with high caliber.
MATERIALS AND METHODS
Location and duration of study: The present study was carried at the Department of Genetics and Biotechnology Laboratory, Faculty of Science, Osmania University, Hyderabad, India, from July 2018 to September 2019.
Steps in homology modeling: The evaluation of the homology model is a multi-step process that can be summarized in following way (1) Template for Identification, (2) Alignments for single or multiple sequences, (3) Target based on the 3D structure of the template for model building, (4) Model refinement, alignments analysis, gap additions and deletions and (5) Model validation
Homology modeling: Homology modeling is relative modeling depends on the natural truth that when two groupings share high closeness/personality, their particular structures are likewise comparative10. In protein 3D structure expectation technique that requirements less time and lower cost with clear advances. In this manner, homology modeling is generally utilized for the generation of 3D structures of proteins with high caliber. This has changed the methods for docking and virtual screening strategies that depend on the structure in the medication disclosure process.
In this strategy, the 3D structure of a protein is acquired with the accompanying advances (I) for a given objective succession, distinguishing the best possible layout utilizing BLAST search, (ii) grouping arrangement (iii) arrangement adjustments to guarantee the moderated or practically significant build-ups are adjusted, (iv) spine generation (v) circle modeling, (vi) chain modeling utilizing rotamer libraries, (vii) improving the model utilizing vitality minimization and (viii) approving the model by stereo-substance assessment utilizing the deposits in the permitted areas of Ramachandra plot just as good energies. Modeler is one of the broadly utilized computational technics for anticipating protein 3D structures utilizing homology modeling11.
Template (fold) recognition and alignment: This is the underlying advance wherein the program/server contrast the grouping of obscure structure and realized structure put away in PDB. The most mainstream server is BLAST (Basic Local Alignment Search Tool) A search with BLAST against the database for ideal local alignments with the question, give a rundown of realized protein structures that matches the grouping12. BLAST can't discover a layout when the grouping character is well underneath 30%; homology hits from BLAST are not dependable. The grouping alignment is progressively touchy in recognizing transformative connections among proteins and genes. The subsequent profile succession alignment appropriately adjusts around 42-47% of build-ups in the 0-40% grouping character extend, this number is roughly twofold than that of the pair shrewd arrangement techniques. Alignment errors are the primary driver of deviations in relative modeling notwithstanding when the right format is picked. As of late, huge advancement has been made in the improvement of touchy alignment strategies dependent on iterative searches, for example, PSI-BLAST, Hidden Markov Models (HMM), for example, SAM, HMMER or profile alignment, FFAS03, profiles can and HH search (Table 1). Numerous alignments are normally heuristic surely understood as dynamic alignment (Fig. 1). Dynamic alignments are easy to perform and permit huge alignments of remotely related successions to be developed. This is actualized in the most generally utilized projects (Clustal W and Clustal X). Alignment of disparate protein successions can be performed with high precision utilizing Clustal W program.
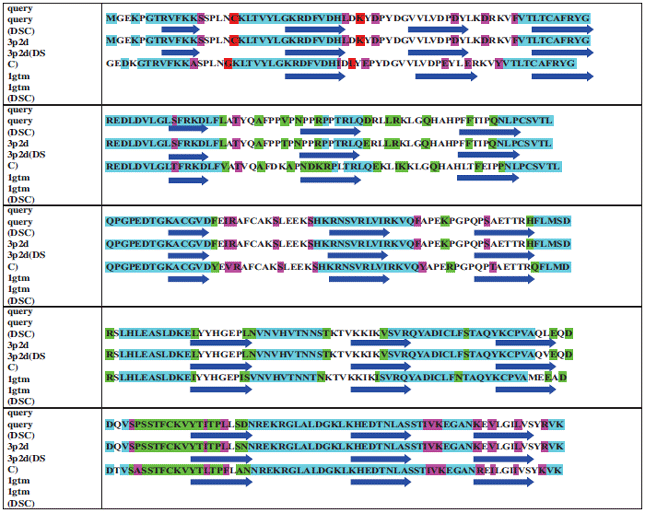 | |
| Fig. 1: | β-Arrest in is a family member of Multiple sequence alignment and query is experimentally derived sequence taken from UNIPROT (ID: P32121) aligned with sequences of PDB entry codes of 3P2D and 1G4M, Discovery Studio Visualizer 2.5 is using for identical residues and conserved residues are indicated in the form of secondary structure |
| Table 1: | Sequence alignment programs and their web server sites |
 | |
Clustal W incorporates numerous highlights like appointing individual loads to each succession in an incomplete alignment and amino acid substitution networks are changed at various alignment stages as indicated by the dissimilarity of the arrangements to be adjusted. Explicit significance is given to build up explicit whole punishments in hydrophilic locales which energize new holes in potential circle districts. Well, are a class of probabilistic models that are generally pertinent to time arrangement or straight succession13. Profile HMM is exceptionally compelling in distinguishing saved examples in numerous groupings.
Modeling building with artificial evolution: The alignment between the question and the layout can be considered as a rundown of activities, for example, build-up transformation, addition or erasure. Assume the layout is the "parent structure", it would take Nature billions of years to develop the format to the objective14. Every activity, i.e., change, cancellation or addition, will bother the format structure and accordingly include a vitality cost, either positive or negative15 the model structure begins from the task with the least vitality cost, etc. The request for the first-round activity does not need to be dictated by really computing the vitality cost for every task; rather, it very well may be helpfully assessed from an experimental perspective16. For instance, transformation is generally simpler in advancement than inclusion and erasure. Accordingly, transformation activities on build-ups that are on the protein surface are normally performed first pursued by change on covered little measured deposits, etc.17. Inclusion or cancellation of different deposits is considered as a gathering of activities, each working on one build up. Model precision depends on the format choice and alignment exactness18. Modeling by fulfillment of spatial limitations dependent on the generation of numerous requirements or restrictions on the structure of objective grouping19.
Loop modeling: Homologous proteins have holes or additions in arrangements, alluded to as loops whose structures are not rationed during development. Loops are considered as the most factor areas of a protein where inclusion and cancellation frequently happen. Loops frequently decide the practical explicitness of a protein structure. Loops add to dynamic and restricting locales. The exactness of loop modeling is the main consideration in deciding the value of homology models for examining ligand associations20. Loop structures are harder to anticipate than the structure of the geometrically profoundly normal strands and helices since loops display more prominent auxiliary changeability than strands and helices of a loop district is generally a lot shorter than that of the entire protein chain. Modeling a loop district has difficulties, which are not liable to be available in the worldwide protein structure. Displayed loop structure must be geometrically predictable with the remainder of the protein structure (Table 2).
Loop prediction: The basic objective is to foresee the conformation of a loop that is fixed at the two finishes by the protein spine. Loop prediction is frequently viewed as a small scale protein collapsing issue. The methodologies fall into two principal classifications: abdominal muscle initio (once more) and database techniques. When loops are generated along these lines, lively criteria (or arrangement likeness for database approach) are regularly connected to choose the in all probability applicant21. Significantly, close local conformations be available among the up-and-comer conformations generated in the initial step of loop modeling. For loops of under 12 buildups, sufficient examining does not seem, by all accounts, to be an issue. Regardless of whether the Protein Data Bank has been altogether expanded from that point forward, late research demonstrated that the database search technique is overwhelmed by the abdominal muscle beginning strategy at around six build-ups loop length22.
| Table 2: | Loop modeling program |
 | |
The precision of loop modeling is profoundly needy not just on the number of build-ups on the up and up, yet besides on the separation between the loop stems.
Side-chain modeling: Side-chain modeling is a remarkable advance in predicting protein structure by homology. The side-chain quality can recognize the portion of right rotamers found or divided by root mean square deviation (RMSD) for all molecules23. The significant bit of leeway is to increase computing effectiveness, since awful rotamers, for example clashing with the spine, have been consequently expelled during the development of the rotamer library. Cook and his collaborators have built up a "solvated rotamer" approach that shows enhancement for side-chain packing at the protein-protein interface. This methodology broadens flow side-chain packing techniques by using a rotamer library including solvated rotamers with at least one water particles fixed to polar utilitarian gatherings in likely hydrogen bond directions, together with a basic vigorous depiction of water-interceded hydrogen bonds.
Other advances in model refinement: Ongoing endeavors on model refinement have been mainly accomplished from increasing alignment exactness. Practically all alignment programming as of now in use needs to depend on one of the derivatives of dynamic programming. Likewise, a genetic algorithm is additionally a significant tool to increase model quality dependent on numerous layouts. The numerous models, each dependent on one format, will be superimposed by Greer24. Variable locales distinguished are traded and improved among various models. In the enhancement procedure, a rmsd constraint can be connected to limit sampling to the conformational space near the found the middle value of the system of the original formats.
RESULTS AND DISCUSSION
Model validation: The most significant factor in the appraisal of developed models is scoring capacity. The projects assess the area of every build-up in a model regarding the normal condition as found in the high-goals X-beam structure. Methods used to determine mist reading in X-beam structures can be utilized to determine alignment errors in homology models by Levitt25. Errors in the model are particularly normal and most consideration is required towards refinement and validation. Errors in the model are typically evaluated by (1) superposition of model onto the local structure with the structure alignment program structural and estimation of RMSD of Cα molecules; (2) generation of Z-score, a proportion of factual centrality between coordinated structures for the model, using the structure alignment program CE, scores four indicate great auxiliary likeness and (3) advancement of a scoring capacity that is fit for discriminating great and awful models. The Ramachandran plot is presumably the most dominant determinant of the nature of the protein, when Ramachandran plot nature of the model is similarly more terrible than that of the format, at that point all things considered, the mistake occurred in spine modeling. Conformational free vitality distinguishes the local structure of a protein from an incorrectly collapsed distraction. A distinct preferred position of such physically inferred capacities is that they depend on all around defined physical interactions, hence making it simpler to take in and to gain insight from their performance. What's more, abdominal muscle initio strategies indicated achievement in late CASP. One of the real downsides of physical concoction portrayal of the folding free vitality of a protein is that the treatment of solvation required normally comes at a critical computational cost. Quick solvation models, for example, the generalized conceived and an assortment of disentangled scoring plans may demonstrate to be very valuable in such manner. Various uninhibitedly accessible projects can be utilized to check homology models, among them WHAT CHECK takes care of regularly crystallographic issues. The validation projects are generally of two sorts: (1) first class (for example PROCHECK and WHATIF) checks for appropriate protein stereochemistry, for example, symmetry checks, geometry checks (chirality, bond lengths, bond edges, torsion edges models, solvation) and basic packing quality and (2) the subsequent classification (e.g. VERIFY3D and PROSAII) check the wellness of succession to structure and doles out a score for every build-up fitting its present condition. GRASP2 is new model appraisal programming created by Honig.
| Table 3: | Model assessment and validation program |
 | |
For instance, holes and insertions can be mapped to the structures to check that they bode well geometrically. It is proposed that manual inspection ought to be combined with existing projects to further distinguish issues in the model (Table 3).
Opportunities and possible challenges in homology modeling: Homology modeling focuses on building 3D structure of proteins from their sequences by using templates with accuracy which is similar to the experimental methods. So, it has an important role in filling the broad gap. Homology modeling applications generally use in drug discovery need high- quality models. As a result, good side-chain modeling, loop modeling and high sequence similarity are crucial in determining further applications of the model build in the drug discovery process.
The number of high-quality protein 3D structures has developed in the last decades. The introduction of new experimental methods like cryo-electron microscopy (Cryo-EM) is anticipated to increase the number of 3D structures resolved experimentally. As the experimentally determined number of high- quality 3D protein structures of protein families increases, the role of homology modeling in determining the 3D structures of the rest of the sequences in these families increases. Although, 3D structures of all protein distinct folds in nature have not been completed yet. As a result, there are some difficulties in building 3D structures of proteins in which the structures of their protein families have not been determined26. The side-chain quality can be divided by root mean square deviation (RMSD) for all molecules. The constructed models are using with various parameters; Template Modeling (TM) score; discrete optimized protein energy (DOPE) score and RMSD value are using for comparison and find the best model. The determined parameter depends on the purpose of modeling results27. The accurate alignment explains that a meaningful model can be generated by every sequence for which a suitable template exists28.
Multiple Sequence Alignments (MSAs) have a primary role in several domains of modern molecular and bioinformatics such as protein 3D structure (or) function prediction, phylogeny inference, molecular function, intermolecular interactions and many other common tasks in sequence analysis. BAliBASE was the first database of model alignments specifically developed to explore the accuracy of the MSA tool. Several studies were reported about the accuracy and efficiency of MSA tools but their focus was individual MSA tools but less effort was made to study the underlying algorithms29. The quantity of high-quality protein 3D structures has increased in the most recent decades. The introduction of new exploratory methods like Cryo-electron microscopy (Cryo-EM) is foreseen to increase the quantity of 3D structures determined tentatively. As the tentatively determined number of high quality 3D protein structures of protein families increases, the job of homology modeling in determining the 3D structures of the remainder of the groupings in these families increases. Nonetheless, 3D structures of all protein distinct overlays in nature have not been finished at this point. Accordingly, there are a few troubles in building 3D structures of proteins wherein the structures of their protein families have not been determined by Rost30. There are many methods utilized for model building in homology modeling. Using the BLAST algorithm instead of typical BLAST may give ideal layout choices in developmental far off cases. New methods with new algorithms have been created. Different examinations have exhibited that there is no single modeling project or server which is better in each property than others. Along these lines, selecting the technique/s to be utilized according to the protein close by and explicit point of future utilizations of the model is significant. For instance, there are attempts to integrate modeling tools with thermo stabilizing changes. Homology modeling may leave some uncertain inquiries in the computational models. The improvement includes refinement of the generated models with atomic elements reproductions by Xiang31. A huge number of models can be worked in a brief timeframe with Linux clusters, each dependent on one variety of alignment. A powerful scoring vitality can be promptly connected to the gathering of models. These models can be additionally minimized with a methodology like genetic algorithm, i.e., shuffling portions among various models by fixing other pieces of protein, where the stems of the section ought to have indistinguishable deposits lined up with the layout. In the present scenario, there are many advances in the tools and servers of homology modeling that improve the accuracy of modeling results and it has an impact on each step of homology modeling in drug design.
CONCLUSION
Protein structure assumes a key job in understanding the component of protein work. Tentatively solving protein structure is a dull procedure that cannot fulfill the need came about because of the exponential growth of protein groupings. Protein structure prediction has been a fantasy for an academic network for decades, for computational scientific experts, yet also for physicist, mathematician and PC researcher. The fantasy has not been seen attainable up to this point with the blast of arrangement and auxiliary information and because of computational advances in a wide range of zones including succession profile investigation and a better understanding of vigorous determinants of protein dependability. In the following 10 years, a basic genomics venture could guide out all the distinct overlap in Nature, which makes it conceivable to tackle the issue of protein structure prediction dependably using homology modeling strategy.
SIGNIFICANCE STATEMENT
The significance of homology modeling has been relentlessly expanding a result of the enormous hole that exists between the staggering number of accessible protein arrangements and tentatively illuminated protein structures and more critically, on account of the expanding unwavering quality and precision of the technique. The on-going advances in homology modeling that improve the accuracy of modeling results and it has an impact on each step of homology modeling, especially in recognizing and adjusting successions to layout structures, far off homologs, modeling of loops and side chains just as identifying mistakes in a model added to the reliable forecast of protein structure, which was impractical even quite a long while back and these studies would helpful for homology in the prediction of protein structure. Protein 3D structure technique needs less time and lower cost with more advances. The high sequence similarity an outstanding advance in predicting protein structure by homology challenges and other computer-aided drug design methods are also expected in future drug discovery.
ACKNOWLEDGMENT
Author acknowledges to Department of Genetics and Biotechnology Laboratory, Osmania University, Hyderabad, India for technical support.
REFERENCES
- Cavasotto, C.N. and S.S. Phatak, 2009. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today, 14: 676-683.
CrossRefDirect Link - Floudas, C.A., H.K. Fung, S.R. McAllister, M. Monnigmann and R. Rajgaria, 2006. Advances in protein structure prediction and de novo protein design: A review. Chem. Eng. Sci., 61: 966-988.
CrossRefDirect Link - Oshiro, C., E.K. Bradley, J. Eksterowicz, E. Evensen and M.L. Lamb et al., 2004. Performance of 3d-database molecular docking studies into homology models. J. Med. Chem., 47: 764-767.
CrossRefDirect Link - Mukhopadhyay, A., N. Borkakoti, L. Pravda, J.D. Tyzack, J.M. Thornton and S. Velankar, 2019. Finding enzyme cofactors in protein data bank. Bioinfo., 35: 3510-3511.
CrossRefDirect Link - Chandonia, J.M. and S.E. Brenner, 2005. Implications of structural genomics target selection strategies: Pfam5000, whole genome and random approaches. PROTEINS: Struct. Function Bioinform., 58: 166-179.
CrossRefDirect Link - Kundrotas, P.J., M.F. Lensink and E. Alexov, 2008. Homology-based modeling of 3D structures of protein-protein complexes using alignments of modified sequence profiles. Int. J. Biol. Macromolecules, 43: 198-208.
CrossRefDirect Link - Barril, X. and S.D. Morley, 2005. Unveiling the full potential of flexible receptor docking using multiple crystallographic structures. J. Med. Chem., 48: 4432-4443.
CrossRefDirect Link - Vitkup, D., E. Melamud, J. Moult and C. Sander, 2001. Completeness in structural genomics. Nat. Struct. Mol. Biol., 8: 559-566.
CrossRefDirect Link - Gupta, A., P. Mohanty and S. Bhatnagar, 2016. Protein structure prediction using homology modeling. IGI Global, 14: 339-359.
CrossRefDirect Link - Tramontano, A. and V. Morea, 2003. Assessment of homology-based predictions in CASP5. Proteins, 53: 352-368.
CrossRefDirect Link - Westbrook, J., Z. Feng, L. Chen, H. Yang and H.M. Berman, 2003. The protein data bank and structural genomics. Nucleic Acids Res., 31: 489-491.
CrossRef - Johnson, M.S., N. Srinivasan, R. Sowdhamini and T.L. Blundell, 1994. Knowledge-based protein modeling. Critical Rev. Biochem. Mol. Biol., 29: 1-68.
CrossRefDirect Link - Lesk, A.M. and C.H. Chothia, 1986. The response of protein structures to amino-acid sequence changes. Phil. Trans. R. Soc. Lond. A, 317: 345-356.
CrossRefDirect Link - Marti-Renom, M.A., A.C. Stuart, A. Fiser, R. Sanchez, F. Melo and A. Sali, 2000. Comparative protein structure modeling of genes and genomes. Ann. Rev. Biophys. Biomol. Struct., 29: 291-325.
CrossRefPubMedDirect Link - Katz, B.A., P.A. Sprengeler, C. Luong, E. Verner and K. Elrod et al., 2001. Engineering inhibitors highly selective for the S1 sites of Ser190 trypsin-like serine protease drug targets. Chem. Biol., 8: 1107-1121.
CrossRefDirect Link - Sadreyev, R.I. and N.V. Grishin, 2004. Quality of alignment comparison by COMPASS improves with inclusion of diverse confident homologs. Bioinfo., 20: 818-828.
CrossRefDirect Link - Notredame, C., D.G. Higgins and J. Heringa, 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol., 302: 205-217.
CrossRefPubMedDirect Link - Shi, J., T.L. Blundell and K. Mizuguchi, 2001. FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure- dependent gap penalties. J. Mol. Biol., 310: 243-257.
CrossRefPubMedDirect Link - Do, C.B., M.S.P. Mahabhashyam, M. Brudno and S. Batzoglou, 2005. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res., 15: 330-340.
CrossRefDirect Link - Katoh, K., K.I. Kuma, H. Toh and T. Miyata, 2005. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res., 33: 511-518.
CrossRefDirect Link - Thompson, J.D., F. Plewniak and O. Poch, 1999. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res., 27: 2682-2690.
CrossRefPubMedDirect Link - Collura, V., J. Higo and J. Garnier, 1993. Modeling of protein loops by simulated annealing. Protein Sci., 2: 1502-1510.
CrossRef - Browne, W.J., A.C. North, D.C. Phillips, K. Brew, T.C. Vanaman and R.L. Hill, 1969. A possible three-dimensional structure of bovine lactalbumin based on that of hen's egg-white lysosyme. J. Mol. Biol., 42: 65-86.
CrossRefPubMedDirect Link - Greer, J., 1990. Comparative modelling methods: application to the family of the mammalian serine proteases. Proteins., 7: 317-334.
CrossRefDirect Link - Levitt, M., 1992. Accurate modeling of protein conformation by automatic segment matching. J. Mol. Biol., 226: 507-533.
CrossRefDirect Link - Muhammed, M.T. and E. Aki-Yalcin, 2019. Homology modeling in drug discovery: Overview, current applications and future perspectives. Chem. Biol. Drug Design, 93: 12-20.
CrossRefDirect Link - Nayeem, A., D. Sitkoff and S. Krystek, 2006. A comparative study of available software for high-accuracy homology modeling: From sequence alignments to structural models. Protein Sci., 15: 808-824.
CrossRefDirect Link - Kuziemko, A., B. Honig and D. Petrey, 2011. Using structure to explore the sequence alignment space of remote homologs. PLoS Comput. Biol., Vol. 7.
CrossRefDirect Link - Babar, M.E., M.T. Pervez, A. Nadeem, T. Hussain and N. Aslam, 2014. Multiple sequence alignment tools: Assessing performance of the underlying algorithms. J. Appl. Environ. Biol. Sci., 4: 76-80.
Direct Link - Rost, B., 1999. Twilight zone of protein sequence alignments. Protein Eng., 12: 85-94.
CrossRefPubMedDirect Link - Xiang, Z., 2006. Advances in homology protein structure modeling. Curr. Protein Peptide Sci., 7: 217-227.
CrossRefDirect Link