
Ameer Khusro
Research Department of Plant Biology and Biotechnology, Loyola College, 34 Chennai, Tamil Nadu, India
LiveDNA: 91.8089
Chirom Aarti
Research Department of Plant Biology and Biotechnology, Loyola College, 34 Chennai, Tamil Nadu, India
Paul Agastian
Research Department of Plant Biology and Biotechnology, Loyola College, 34 Chennai, Tamil Nadu, India
Trends in Bioinformatics
Year: 2020 | Volume: 13 | Issue: 1 | Page No.: 1-9
DOI: 10.3923/tb.2020.1.9







ABSTRACT
Background and Objective: At present, millions of mortalities caused by tuberculosis and cancer are of colossal concern worldwide. The severe adverse effects of existing drugs have emphasized the researchers to identify ideal therapeutic agents. In view of this, the present study was aimed to assess the anti-tubercular and anticancer properties of bacterial peptide using in silico docking tool. Materials and Methods: The 3D structure of peptide NMANF2 was modelled using online software PEP-FOLD and iCn3D. The 3D structures of M. tuberculosis, lung cancer (A540) and colon cancer (HT-29) cells’ target receptors were retrieved from RCSB PDB. In silico molecular docking between ligands and targeted proteins of M. tuberculosis and cancer cell lines (A540 and HT-29) were analyzed and visualized using Hex 8.0.0 docking software. Results: The peptide exhibited highest negative energy value (E-value) with DNA gyrase, followed by ribonucleotide reductase, LysA, alanine racemase and isocitrate lyase of M. tuberculosis. The peptide revealed highest docking score with Bcl-2 of A540 cell line. On the other hand, the peptide showed highest negative E-value with CDK4 among targeted proteins of HT-29 cell line. Conclusion: Based on this in silico results, peptide NMANF2 may be used as potent agent for designing anti-tubercular and anticancer drugs in future.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Ameer Khusro, Chirom Aarti and Paul Agastian, 2020. In silico Modelling and Molecular Docking Insight of Bacterial Peptide for Anti-tubercular and Anticancer Drug Designing. Trends in Bioinformatics, 13: 1-9.
DOI: 10.3923/tb.2020.1.9
URL: https://scialert.net/abstract/?doi=tb.2020.1.9
DOI: 10.3923/tb.2020.1.9
URL: https://scialert.net/abstract/?doi=tb.2020.1.9
INTRODUCTION
Currently, the lengthy therapies and side effects of existing drugs have emphasized the worldwide researchers to search for promising anti-tubercular and anticancer agents. Among disparate therapeutic agents, peptides have created colossal interest due to their pivotal molecular characteristics and unique mode of actions1. Bacteria associated therapeutic peptides are ubiquitous in nature and have been investigated extensively in the past few decades for designing ideal antibiotics2. Peptides are generally composed of short amino acids residues constituting small cationic and amphiphilic moieties. Based on the composition and structures, peptides have been grouped into distinct classes, possessing α-helical structure (α family), β-strands (β family), both α-helical and β-strands (α β family) and neither α-helical structure nor β-strands (non-α β family)3.
Plethora of studies has demonstrated that peptides could have many applications in medicine due to broad range of biological targets, thereby exhibiting remarkably high activity and potentially reduced toxicity than other small molecules4. Additionally, some peptides demonstrate multiple modes of action, which can be effective in improving biological potency and evading resistance process of pathogens5. Therefore, a number of research activities have focused on the diverse applications of peptides towards the discovery of novel therapeutic drugs.
Naturally-occurring peptides often possess diversified attributes and indicate pronounced class of lead components for designing new antibiotics1. Peptide properties viz. charge, hydrophobicity, primary and secondary structure, structure and interaction with targets are leading parameters in the prediction of peptide to be pivotal therapeutic agents6.
Peptide-protein interactions are important in several cellular functions. Hence, determination of the structure of peptide-protein complexes is crucial in order to understand the molecular mechanism of related biological processes and developing peptide-based drugs. The interaction between peptide and protein play a pivotal role in distinct biological processes such as signal transduction, immune responses and cellular regulation7. About 40% of the protein-protein interactions are mediated by short peptides1,2. Hence, determining the structure of peptide-protein complexes is important for understanding the molecular process and thus modulating the protein-protein interactions for therapeutic purposes8. Now-a-days, very few peptide-protein complex structures are experimentally implied due to the high cost and technical difficulties in in vitro studies. Therefore, molecular docking has been widely utilized towards the determination of peptide-protein complex structures. Peptide-protein docking predicts the complex structure by sampling plausible peptide binding conformations with an energy scoring function7.
Computational simulations or approaches are important for selecting therapeutic peptides as the hypothesis can be tested prior to the time-consuming and resource-demanding synthesis. In view of this, the present study was aimed to design peptide-based drugs against tuberculosis (TB) and cancer (lung and colon cancer cell lines) by means of in silico studies to aid efforts towards novel therapeutic strategy.
MATERIALS AND METHODS
Location and duration of study: The present study was carried in Department of Plant Biology and Biotechnology, Loyola College, Chennai, India from April, 2019 to mid of October, 2019.
Peptide of interest: Peptide NMANF2, previously isolated from Staphylococcus hominis9 strain MANF2, was used in this investigation. The amino acid residues of peptide NMANF2 were determined as ‘KAIGLVIPEIDGKLDGGAQRV’ .
Structure modelling of peptide: The 3D conformations of peptide (21 amino acid residues) were predicted using online software PEP-FOLD10 and iCn3D (https://www.ncbi.nlm.nih. gov/Structure/icn3d/full.html).
Molecular docking
Ligands selection: The structure of peptide NMANF2 obtained using PEP-FOLD (http://bioserv.rpbs.univ-paris-diderot.fr/ services/PEP-FOLD3/) was saved as PDB (Protein Data Bank) file and used for molecular docking. On the other hand, structure of rifampicin (anti-tubercular drug) and doxorubicin (anticancer drug) were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and were used for docking analysis too.
Targeted Mycobacterium tuberculosis proteins: The 3D structures of receptors viz. alanine racemase, DNA gyrase, isocitrate lyase, LysA and ribonucleotide reductase were retrieved from RCSB PDB (http://www.rscb.org/pdb). The complexes bound to the receptor, such as non-essential water molecules and any inhibitors were removed while docking.
Targeted lung and colon cancer proteins: For targeting lung cancer (A540 cell line), the 3D structures of Bcl-2 (B cell lymphoma/leukemia type 2), H-Ras and epidermal growth factor receptor (EGFR) tyrosine kinase were retrieved from RCSB PDB (http://www.rscb.org/pdb). The complexes bound to the receptor, such as non-essential water molecules and inhibitors were removed from the target receptors while docking.
Likewise, for targeting colon cancer (HT-29 cell line), Bcl-xL (B-cell lymphoma-extra large), CDK2 (cyclin dependent kinase 2) and CDK4 (cyclin dependent kinase 4) were retrieved from RCSB PDB (http://www.rscb.org/pdb). The complexes bound to the receptor, such as non-essential water molecules and inhibitors were removed from the target receptors while docking.
Peptide-protein docking and visualization: Molecular docking between ligands and targeted proteins of M. tuberculosis and cancer cell lines (A540 and HT-29) were analyzed and visualized using Hex 8.0.0 docking software. Hex is an interactive molecular graphics program that reads in molecular coordinate files and displays in silico interaction in varied representations and colour schemes. The tool identifies the ligand with the best score and calculates the ligand-receptor interaction with the lowest free energy value.
The docking was performed by adjusting following parameters/features.
| • | Correlation type-shape+electrostatics |
| • | FFT mode-3D |
| • | Post processing-MM energies |
| • | Grid dimension-0.6 |
| • | Receptor range-180 |
| • | Ligand range-180 |
| • | Twist range-360 |
| • | Distance range-40 |
The binding energy (KJ moL–1) estimated after docking was tabulated.
RESULTS
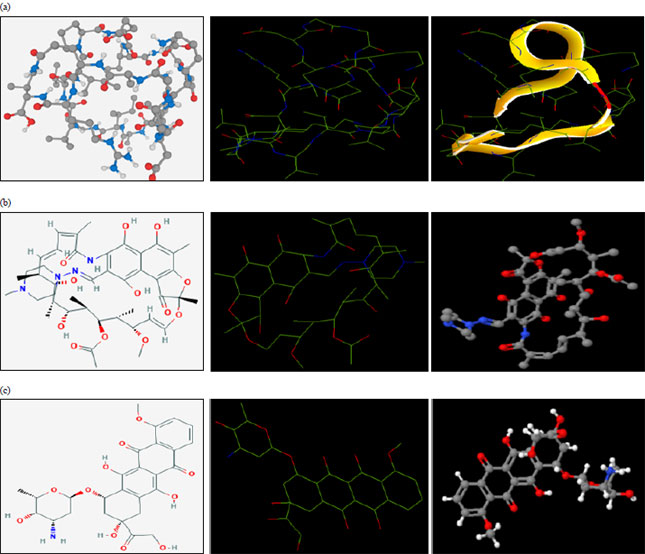
Structural modelling of peptide: Figure 1 shows modelled predicted structures such as ‘Lines’, ‘Cartoon’, ‘Ribbon’, ‘Ball and Stick’, ‘Sphere’, ‘Stick and Surface’ of peptide NMANF2, as determined by PEP-FOLD and iCn3D online tool.
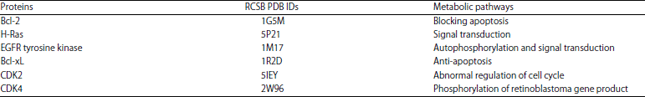
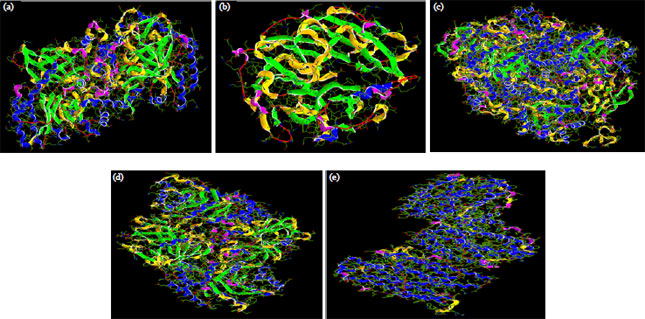
In silico molecular docking: Structures of ligands used (peptide NMANF2, rifampicin and doxorubicin) are shown in Fig. 2. The RCSB PDB IDs and metabolic pathways of M. tuberculosis receptors are illustrated in Table 1. Likewise, RCSB PDB IDs and metabolic pathways of cancer cells receptors are described in Table 2.
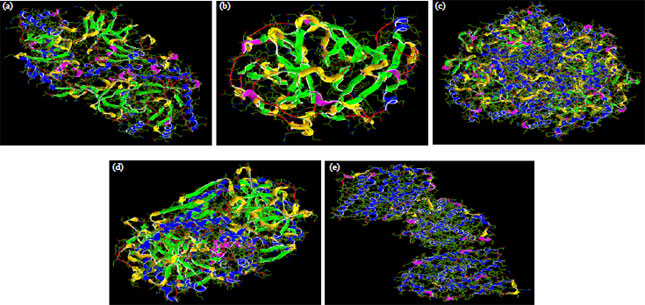
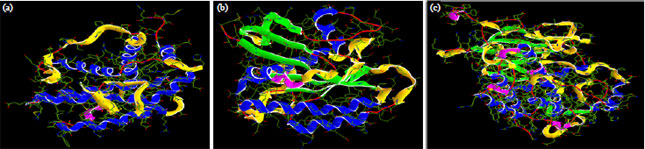
Table 3 shows the binding affinity between peptide NMANF2 and targeted proteins of M. tuberculosis using Hex 8.0.0 docking software. The peptide exhibited highest negative energy value (E-value) of -635.15 KJ moL–1 with DNA gyrase, followed by -581.79, -551.88, -504.75 and -79.34 KJ moL–1 with ribonucleotide reductase, LysA, alanine racemase and isocitrate lyase respectively (Fig. 3). Furthermore, rifampicin showed E-values with M. tuberculosis proteins in the order of alanine racemase (-332.23 KJ mol–1)> ribonucleotide reductase (-324.09 KJ moL–1)>LysA (-316.08 KJ moL–1)>DNA gyrase (-300.01 KJ moL–1) > isocitrate lyase (-248.83 KJ moL–1) (Table 3, Fig. 4).
| Table 1: | RCSB PDB IDs and metabolic pathways of M. tuberculosis targeted proteins |
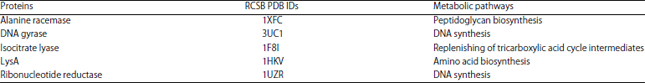 | |
| Table 2: | RCSB PDB IDs and metabolic pathways of A540 and HT-29 cancer cells proteins |
 | |
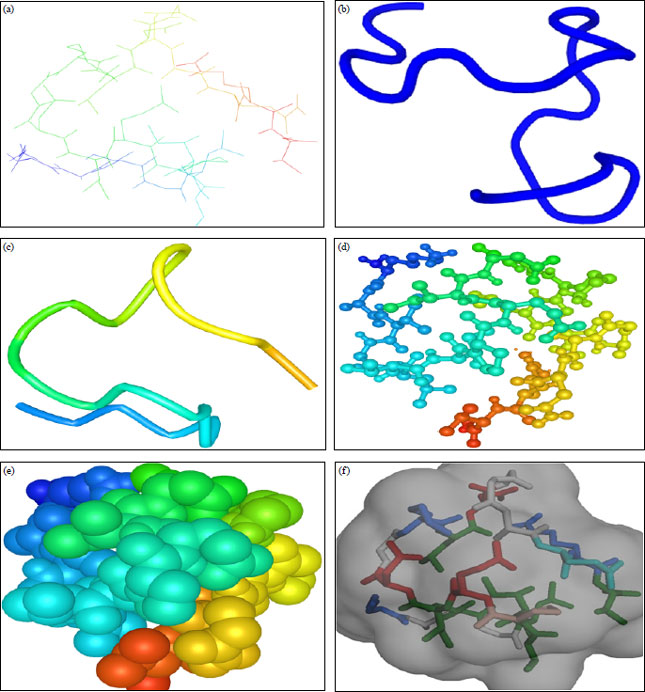 | |
| Fig. 1(a-f): | Structures of peptide NMANF2 obtained using PEP-FOLD and iCn3D, (a) Lines, (b) Cartoon, (c) Ribbon, (d) Ball and stick, (e) Sphere and (f) Stick and surface |
| Table 3: | Docking analysis of peptide NMANF2 and rifampicin with M. tuberculosis receptors |
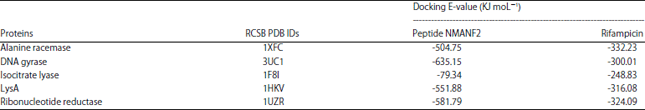 | |
 | |
| Fig. 2(a-c): | Different structures of ligands, (a) Peptide NMANF2, (b) Rifampicin and (c) Doxorubicin |
| Table 4: | Docking analysis of peptide NMANF2 and doxorubicin with A540 and HT-29 cancer cells receptors |
 | |
Among targeted proteins of A540 cell lines, peptide NMANF2 revealed highest docking score of -470.6 KJ moL–1 with Bcl-2 (Table 4, Fig. 5). On the other hand, the peptide showed highest negative E-value of -378.9 KJ moL–1 with CDK4 among targeted HT-29 cell lines (Table 4, Fig. 6). Molecular docking of doxorubicin with selected proteins of A540 and HT-29 cancer cells is shown in Fig. 7 and 8, respectively. Doxorubicin revealed highest docking score of -288.36 and -308.7 KJ moL–1 with Bcl-2 and CDK4, respectively (Table 4).
 | |
| Fig. 3(a-e): | Molecular docking of peptide NMANF2 with, (a) Alanine racemase, (b) DNA gyrase, (c) Isocitrate lyase, (d) LysA and (e) Ribonucleotide reductase |
Peptide exhibited E-value of -504.75, -635.15, -79.34, -551.88 and -581.79 KJ moL–1 with alanine racemase, DNA gyrase, isocitrate lyase, LysA and ribonucleotide reductase, respectively | |
 | |
| Fig. 4(a-e): | Molecular docking of rifampicin with, (a) Alanine racemase, (b) DNA gyrase, (c) Isocitrate lyase, (d) LysA and (e) Ribonucleotide reductase |
| Rifampicin exhibited E-value of -332.23, -300.01, -248.83, -316.08 and -324.09 KJ moL–1 with alanine racemase, DNA gyrase, isocitrate lyase, LysA and ribonucleotide reductase, respectively | |
 | |
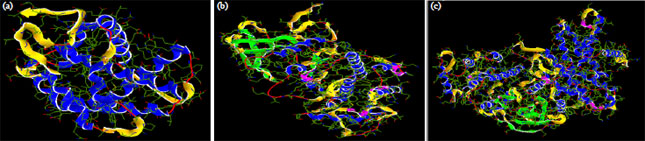
| Fig. 5(a-c): | Molecular docking of peptide NMANF2 with, (a) Bcl-2, (b) H-Ras and (c) EGFR tyrosine kinase of A540 cell line |
Peptide showed E-value of -470.6, -416.78 and -417.63 KJ moL–1 with Bcl-2, H-Ras and EGFR tyrosine kinase, respectively | |
 | |
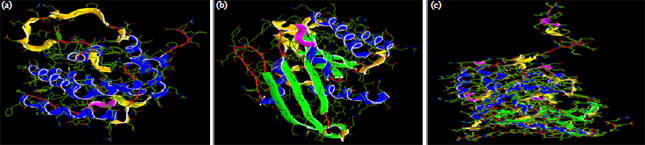
| Fig. 6(a-c): | Molecular docking of peptide NMANF2 with, (a) Bcl-xL, (b) CDK2 and (c) CDK4 of HT-29 cell line |
Peptide showed E-value of -374.83, -353.62 and -378.9 KJ moL–1 with Bcl-xL, CDK2 and CDK4, respectively | |
 | |
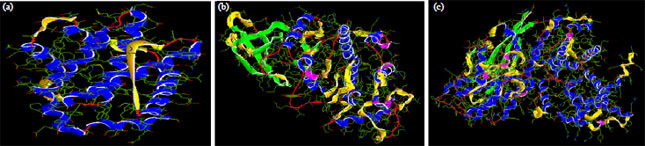
| Fig. 7(a-c): | Molecular docking of doxorubicin with, (a) Bcl-2, (b) H-Ras and (c) EGFR tyrosine kinase of A540 cell line |
Doxorubicin showed E-value of -288.36, -263.49 and -32.45 KJ moL–1 with Bcl-2, H-Ras and EGFR tyrosine kinase, respectively | |
 | |
| Fig. 8(a-c): | Molecular docking of doxorubicin with, (a) Bcl-xL, (b) CDK2 and (c) CDK4 of HT-29 cell line |
Doxorubicin showed E-value of -275.38, -274.11 and -308.7 KJ moL–1 with Bcl-xL, CDK2 and CDK4, respectively | |
DISCUSSION
In the present study, target proteins of M. tuberculosis were docked with the peptide NMANF2 and rifampicin using Hex 8.0.0 software in order to demonstrate their molecular interactions and binding energy. Peptide NMANF2 exhibited highest negative E-value with DNA gyrase, followed by ribonucleotide reductase, LysA, alanine racemase and isocitrate lyase. Similarly, target proteins of A540 and HT-29 cell lines were docked with peptide NMANF2 and doxorubicin which showed the highest docking score of the peptide with Bcl-2 and CDK4. In this context, peptide NMANF2 and rifampicin were reported as the most potent inhibitors against DNA gyrase and alanine racemase of M. tuberculosis, respectively, thereby showing higher E-values and strong interaction with receptors. Peptide NMANF2 exhibited higher docking score than that of rifampicin. This might be due to the fact that rifampicin had comparatively lower interaction with these selected targeted receptors. Doxorubicin also revealed higher docking score with Bcl-2 and CDK4 of A540 and HT-29 cancer cells. However, the E-values obtained due to the interaction with doxorubicin were reported to be lower than that of peptide NMANF2. This might also be due to the fact that doxorubicin had comparatively lower interaction with these targeted receptors. In complete agreement with the present investigation, recent in silico studies had successfully used Hex 8.0.0 software for evaluating the molecular interaction between ligand of interest and various receptors of diverse diseases11-13. The current findings provided further promising role of Hex 8.0.0 tool in understanding peptide-protein based interaction towards designing ideal drugs against TB and cancer (lung and colon) in future.
In view of the desperate necessity to find new auspicious agents with pronounced anti-tubercular and anticancer agents, the present in silico study predicted the strong role of peptide NMANF2 in designing new therapeutic drugs in future. The insight into the correlation between this peptide structure and its strong binding with target proteins will lead to design novel anti-tubercular and anticancer drugs that might overcome the complications of existing drugs. However, further in vivo study certainly needs to be determined in order to evaluate the non-toxicity attribute of this peptide.
CONCLUSION
In a nutshell, the peptide showed affinity with DNA gyrase, ribonucleotide reductase, LysA, alanine racemase and isocitrate lyase of M. tuberculosis. Similarly, among targeted proteins of A540 cell lines, peptide NMANF2 revealed potential docking with Bcl-2. On the other hand, the peptide was reported to be potential inhibitor of CDK4 among targeted proteins of HT-29 cell line.
SIGNIFICANCE STATEMENT
This in silico study determines the anti-tubercular and anticancer traits of peptide NMANF2 that can certainly be beneficial for designing new peptide-based drugs for TB and cancer treatment in future. This study will help researchers to uncover the potential role of bacterial peptides that many researchers were unable to explore. Thus, this computational approach may emphasize worldwide researchers design ideal peptide-based therapeutic drugs from un/less exploited sources. However, despite effective in silico reports of this study, further in vivo investigations need to be determined in order to analyze efficacy of this peptide as potent drug candidate towards TB and cancer therapy.
ACKNOWLEDGMENT
Authors acknowledge Maulana Azad National Fellowship (F1-17.1/2015-16/MANF-2015-17-BIH-60730), University Grants Commission, Delhi, India for the support.
REFERENCES
- Khusro, A., C. Aarti and P. Agastian, 2016. Anti-tubercular peptides: A quest of future therapeutic weapon to combat tuberculosis. Asian Pac. J. Trop. Med., 9: 1023-1034.
CrossRefDirect Link - Khusro, A., 2016. Therapeutic potential of anti-tubercular peptides: A million dollar question. EC Microbiol., 4: 697-698.
Direct Link - Wang, G., 2013. Database-guided discovery of potent peptides to combat HIV-1 or superbugs. Pharmaceuticals, 6: 728-758.
CrossRefDirect Link - Craik, D.J., D.P. Fairlie, S. Liras and D. Price, 2013. The future of peptide‐based drugs. Chem. Biol. Drug Design, 81: 136-147.
CrossRefDirect Link - Schmitt, E.K., M. Riwanto, V. Sambandamurthy, S. Roggo and C. Miault et al., 2011. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angewandte Chem. Int. Edn., 50: 5889-5891.
CrossRefDirect Link - Juretic, D., D. Vukicevic, N. Ilic, N. Antcheva and A. Tossi, 2009. Computational design of highly selective antimicrobial peptides. J. Chem. Inform. Model., 49: 2873-2882.
CrossRefDirect Link - London, N., B. Raveh and O. Schueler-Furman, 2013. Peptide docking and structure-based characterization of peptide binding: From knowledge to know-how. Curr. Opin. Struct. Biol., 23: 894-902.
CrossRefDirect Link - Fosgerau, K. and T. Hoffmann, 2015. Peptide therapeutics: Current status and future directions. Drug Discov. Today, 20: 122-128.
CrossRefDirect Link - Khusro, A., C. Aarti, A. Dusthackeer and P. Agastian, 2018. Anti-tubercular and probiotic properties of coagulase-negative staphylococci isolated from Koozh, a traditional fermented food of South India. Microb. Pathog., 114: 239-250.
CrossRefDirect Link - Beaufays, J., L. Lins, A. Thomas and R. Brasseur, 2012. In silico predictions of 3D structures of linear and cyclic peptides with natural and non‐proteinogenic residues. J. Peptide Sci., 18: 17-24.
CrossRefDirect Link - Suryawanshi, S.K. and U. Chouhan, 2018. Computational approaches for the prediction of antimicrobial potential peptides from Ocimum tenuiflorum. Asian J. Pharm. Clin. Res., 11: 398-401.
CrossRefDirect Link - Ashwini, S., S.P. Varkey and M. Shantaram, 2017. In silico docking of polyphenolic compounds against caspase 3-Hela cell line protein. Int. J. Drug. Dev. Res., 9: 28-32.
Direct Link - Menakha, M., M. Sangeetha, P. Mani, M.S. Al-Aboody and R. Vijayakumar, 2018. In silico prediction of drug molecule from Ipomoea sepiaria against type 2 diabetes. Prog. Med. Sci., 3: 9-14.
CrossRefDirect Link