
U.S.F. Tambunan
Department of Chemistry, Faculty of Mathematics and Science, University of Indonesia, Depok 16424, Indonesia
R. Harganingtyas
Department of Chemistry, Faculty of Mathematics and Science, University of Indonesia, Depok 16424, Indonesia
A.A. Parikesit
Department of Chemistry, Faculty of Mathematics and Science, University of Indonesia, Depok 16424, Indonesia
Trends in Bioinformatics
Year: 2012 | Volume: 5 | Issue: 2 | Page No.: 25-46







ABSTRACT
In 2009, swine flu attacked various countries in the world. World Health Organization (WHO) set influenza A H1N1 virus disease as a global pandemic on June 11, 2009. At least, there are approximately 18,449 people worldwide who died from this virus attack. Then, on August 10, 2010, WHO officially announced that the swine flu pandemic in the world has ended and changed into post-pandemic phase. The post-pandemic phase is the most appropriate phase to find an antiviral that can overcome the infection with this virus. The existing antivirals, amantadine and rimantadine, are reported to have experienced resistance. Therefore, it is necessary to find a new antiviral to replace amantadine and rimantadine as the M2 channel protein inhibitor of influenza A H1N1 virus. Later, it was reported that compound (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine has the ability to inhibit channel M2 protein of influenza A H1N1 virus. This research modified (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine in silico to obtain better inhibitors. Three inhibitors docking with standard and 52 inhibitor modifications were performed against the M2 protein channel and drug scan for modification inhibitors was also conducted. Docking results had the three best binding affinity of modification inhibitors and its potency of inhibition is much better than the standard ligands. Based on drug analysis scan, the modified inhibitor has good pharmacological properties which are indicated by the value of drug-likeness, drug score, oral bioavailability and toxicity.
PDF Abstract XML References Citation
Received: January 10, 2012;
Accepted: March 20, 2012;
Published: May 24, 2012
How to cite this article
U.S.F. Tambunan, R. Harganingtyas and A.A. Parikesit, 2012. In silico Modification of (1R, 2R, 3R, 5S)-(-)- Isopinocampheylamine as Inhibitors of M2 Proton Channel in Influenza A Virus Subtype H1N1, using the Molecular Docking Approach. Trends in Bioinformatics, 5: 25-46.
DOI: 10.3923/tb.2012.25.46
URL: https://scialert.net/abstract/?doi=tb.2012.25.46
DOI: 10.3923/tb.2012.25.46
URL: https://scialert.net/abstract/?doi=tb.2012.25.46
INTRODUCTION
Influenza A virus is a highly contagious viral pathogen of birds and mammals, including humans (Webby and Webster, 2001). The most infamous pandemic, the Spanish flu, manifested a deep impact on world population mortality. It is estimated that there were at least 50 million mortalities in 1918-1919 (Betakova, 2007). In 2009, swine flu again attacked various countries in the world. World health Organization (WHO) set a pandemic influenza A H1N1 virus as a global pandemic on June 11, 2009. At least, there were approximately 18,449 people worldwide who died from this virus attack. Then, on August 10, 2010, WHO officially announced that the swine flu pandemic in the world has ended and changed into post-pandemic phase. Post-pandemic phase means that the spread of flu activity around the world has returned to a usual level of treatment as it occurs seasonally. Post pandemic phase is a phase most appropriate to find antivirals that can overcome this viral infection. H1N1 virus is a global pandemic, which already affected several countries such as Malaysia, Iran and Nigeria (Yahyapour et al., 2007; Sar et al., 2010; Karim and Razali, 2011). The escalation of this pandemic deserves more concern, because Alsaif et al. (2010) reported that there were certain resistances for taking H1N1 vaccination in Hail Community. This could hamper the process of eradicating the flu pandemic.
Swine flu has been identified as a new strain of influenza virus A H1N1. Influenza A virus H1N1 is a combination of swine influenza virus genes, avian and human. Based on genetic characterization, gene Hemagglutinin (HA) is similar to swine influenza viruses in the United States, while the Neuraminidase gene (NA) and matrix protein (M) is similar to the swine flu virus isolated from Europe. This unique genetic combination has not been previously detected anywhere, and no patient who had direct contact with pigs that could bring the possibility of transmission of this new strain of influenza virus among humans has been seen (http://emedicinemedscape.com).
Many antiviral drugs have been developed to overcome these infections, namely oseltamivir, zanamivir, amantadine and rimantadine, among others. Oseltamivir and zanamivir are neuraminidase inhibitors. Both antiviral drugs are recommended by the Centers of Disease Control and Prevention (CDC) and WHO (Rungrotmongkol et al., 2009), as it is considered effective in treating influenza virus infection that occurs in humans (Lew et al., 2000). However, antiviral resistance has been reported lately. Influenza virus type A (H1N1) resistance against oseltamivir was reported in Europe in years 2007-2008 (Cheng et al., 2009). In addition, there are also amantadines and rimantadine which can be used to inhibit M2 ion-channel proteins (De Clercq, 2006). The U.S. Food and Drug Administration have approved amantadine and rimantadine in October 1966 as an agent for prophylaxis against influenza in Asia. Nonetheless, as time went on, there were possible drug-resistant viruses due to antigenic drift mutations (Beigel et al., 2005; De Jong et al., 2005). The level of amantadine and rimantadine resistance increased from below 10% during 1995-2002, after which it increased to 58% in 2003, 74% in 2004 and 92% during 2005-2006.
M2 channel protein in influenza A virus is one of the targets of anti-influenza drug that had been used to control influenza virus infection, namely, amantadine and rimantadine. However, the recent use of the drugs, amantadine and rimantadine, is limited because of the drugs’ resistance. The resistance due to mutations of the influenza virus strain is a mutation of three residues on influenza A H5N1 virus, that is, at position V28, S31 and L43, the residues were mutated into I28, N31 and T43. Among the three mutated residues, the S31N mutation is most common in the M2 protein channel that can cause resistance to amantadine (Rungrotmongkol et al., 2009). In addition to the resistance of amantadine and rimantadine, it was also recently reported that amantadine also has adverse effects on the central nervous system (De Clercq, 2006).
The harmful amantadine resistance as antiviral drugs was a good motive for searching for inhibitors of the M2 protein channel that can replace the derivative compound of adamantine. Before now, several inhibitors of the M2 protein channel have been reported. They are amantadine, rimantadine, amantadine derivative, inhibitors of non-adamantane, and Isopinocampheylamine (Hu et al., 2010). There are some research groups, such as Sohail et al. (2011), who reported that certain lead compounds from plant could exhibit anti-Influenza-viral resistance. However, their research still needs more conclusive results to comprehend the molecular mechanism of the lead compounds. This is one reason why molecular modeling could play an important role in this field.
This study’s research group has reviewed some notable research on molecular modeling and successfully conducted a research on H5N1 mutation. Moreover, we are confident that our previous publication methodology could be utilized for H1N1 research as well (Tambunan et al., 2008, 2010).
Molecular docking and dynamics are commonly used methods in bioinformatics. Some research groups have successfully utilized it for PB2 of influenza virus, and anti bacterial resistance towards antibiotics (Lakshmi et al., 2011; Amir et al., 2011). Sur et al. (2009) reported that there are high expressions of pathogenicity related genes which confirm its role as pathogen. Moreover, most of the H1N1 basic proteomes are influenced by mutational pressure. This is one reason why proteomic based research on H1N1 is still considered important.
From the research of Zhao et al. (2011), it was reported that compound number 5, the compound (1R, 2R, 3R, 5S)-(-)-isopinocampheylamine, is the most potent compound as inhibitors of the M2 protein channel and is three times more active than the M2 inhibitors’ amantadine as channel protein (IC50 = 1363 vs. 5960 μM). Therefore, this study created a modification of the compound (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine to find a more potent M2 protein channel inhibitor on the H1N1 virus.
The purpose of this research was to design a drug for influenza A H1N1 virus by in silico based compound (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine through molecular docking method. This compound was expected to replace the antivirals amantadine and rimantadine.
MATERIALS AND METHODS
This study was conducted at Bioinformatics Laboratory, Department of Chemistry, Faculty of Mathematics and Science, University of Indonesia. The research duration was between January 2011 and July 2011.
Searching for protein sequences of H1N1 virus M2 channel: The search for protein sequence M2 H1N1 virus in human and pig host server was done by GenBank Influenza Virus (IVR-NCBI) online database. Homepage-IVR servers can be accessed through the NCBI website address at http://ncbi.nlm.nih.gov/genomes/FLU/Database.html. The format sequence used to store the M2 protein sequence was FASTA, because it was the most widely used for genome analysis.
Multiple sequence alignment: H1N1 virus M2 protein sequences of human and pig hosts in FASTA format were incorporated into the display of Multiple Sequence Alignments (MSA) ClustalW2 server. Its homepage can be accessed online at http://www.ebi.ac.uk/Tools/clustalw2/. MSA was performed on the entire H1N1 virus M2 protein. Then, the M2 protein sequences having the highest bit score was used as input for subsequent analysis.
Three-dimensional protein structure search of the M2 channel: The search for three-dimensional (3D) structure of the sequence was carried out using the software of Max Planck Institute Hhpred, which can be accessed through the website http://toolkit.tuebingen.mpg.de/hhpred. The selected 3D structure has the highest similarity to the receptor binding protein M2 channel sequences.
Search data of 3D protein GDP-M2 channel structure: three-dimensional structure of the M2 channel can be downloaded from the PDB database available at the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RSCB-PDB) through http://www.rscb.org/pdb/.
Geometry optimization and energy minimization of M2 protein channel: Geometry optimization and energy minimization of the M2 channel’s 3D structure was performed using Molecular Operating Environment (MOE) software.
Modification of (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine compounds modeled into 3D structures: Modified forms of (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine were performed using ChemSketch software from ACDLabs.
3D structure geometry optimization and energy minimization of ligands: Geometry optimization and energy minimization of the 3D energy structure of (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine (ligand) was performed using MOE software. The algorithm used was the Polak-Ribiere conjugate gradient with a maximum gradient Root Mean Square (RMS) convergence of 0.001 kcal mol-1 Å and molecular mechanics force field parameters MMFFx.
Protein ligand docking with M2 channel: The process begins with the preparation of docking files. It was accomplished using the software contained in the MOE. For both the ligand and the protein, hydrogen was added to both polar and charge forcefield. The M2 channel protein was saved in MOE format for later use in the preparation parameters. Calculations were carried out with the docking parameters of Lamarckian Genetic Algorithm (LGA). These parameters were stored in the MOE format as files used to run the docking process; though the docking process is run using the MOE software.
Determination of protein-ligand complex conformation docking results: The result of docking calculations was saved in text (notepad) format. Determination of protein-ligand conformation docking was done by selecting the ligand conformation that has the lowest binding energy.
Energy association and inhibition constant (Ki): Bond energies and inhibition constants docking results were saved in notepad format. Selected complex protein-ligands which have the smallest value of bond energy and inhibition constants were used for further analysis.
Hydrogen bonding: Hydrogen bonding, which occurs in the M2 channel protein complex best ligand docking results, was identified using MOE software with the input file Mdb.
Residue contacts: Contact residues of complex enzyme-ligand docking results were identified using MOE software.
Analysis of drug scan: Analysis of drug scans was performed on the results of the docking analysis of ligand binding that has a low ΔG0 corresponding to the normal distribution which is 20% of the lowest energy. This analysis was performed by comparing the best ligand docking analysis to the rules of good medicine (Lipinsky's Rule of Five).
Toxicity prediction: The analysis performed on the results of best ligand docking toxicological properties was based on the analysis results of screening and drug scans. Parameters to be seen among others are predicting carcinogenicity and mutagenicity of these ligands by using some software such as ToxTree and Lazar toxicological properties.
RESULTS AND DISCUSSION
Preparation of protein M2 channel of influenza A (H1N1)
Searching for protein sequences of influenza A virus M2 channel (H1N1): Sequences of the M2 protein channel that will serve as the target protein was determined by using a database of influenza contained in the official website of the NCBI (http://www.ncbi.nlm.nih.gov/genomes/flu/). In this site, Influenza Virus Resources Database allows us to get the influenza virus data with the desired specifications. This specification covers the type of influenza, the host of the virus, the isolated virus country of origin, the desired type of protein sequence, and subtypes of influenza viruses. The used parameters are, among others, influenza type A, a selected sequence of the H1N1 subtype M2 protein channel between 2009 and 2010 from the whole world and the human host. Retrieved protein sequences were FASTA format with the full-length type. The purpose of the download was for facilitating the subsequent analysis. From this protein sequences determination, 173 M2 channel sequences from various countries were obtained.
Multiple sequence alignment: One hundred and seventy-three protein sequences of the M2 channel were determined to be targeted using MSA. The resulted best representative sequence will be targeted in this study. The determination was done with online clustalW2 of EBI official site by looking at the similarity score of each compared sequence with the highest value of 100. The selected M2 channel protein sequence that has the highest similarity value (100) is the M2 channel protein of Addis Ababa, Ethiopia, in 2009, with the code matrix protein 2 [Influenza A virus (A/Addis Ababa/WR2848T/2009 (H1N1))] and GenBank code ADM14979.1. NCBI Data which stipulate that sequence> gi | 303385772 | gb | ADM14979.1 | (Attachment 5) was isolated from the human sequence of a 2 year-old girl. These sequences were collected on August 18, 2010 by Watler Reed Army Institute of Research, 503 Robert Grant Ave., Silver Spring, Maryland 20910, USA.
Identification of homology modeling and templates: From the alignment, protein sequence of the M2 channel of Influenza A H1N1 virus (A/Addis Ababa/WR2848T/2009 (H1N1) was used as target proteins for structure determination of 3D template channel M2 protein of influenza A (H1N1). Determination of the 3D structure of the M2 protein channel influenza A H1N1 (A/Addis Ababa/WR2848T/2009 (H1N1) was done using the software of the Max Planck Institute Hhpred, which could be accessed through the website http://toolkit.tuebingen.mpg.de/hhpred. Results of Hhpred showed that the M2 protein channel of Influenza A H1N1 (A/Addis Ababa/WR2848T/2009 (H1N1) has 100% similarity with the M2 channel protein with PDB code 2KIH composed of 43 amino acid residues. The existing crystal structure was the result of Nuclear Magnetic Resonance (NMR) research.
The active visualization of M2 protein channel: 3D structure of the M2 channel protein was obtained from the PDB. Visualization was conducted to see the position of residues that have important functions and will be used as a target residue in the docking process. Based on the publication of Du et al. (2010), it was known that the M2 channel protein activity was determined by three important residues, namely, His37, Trp41 and Asp44, as seen in Fig. 1. Three residues were called the functional residue because each one of them has a special function. They are, His37 as a pH sensor, Trp41 as the channel gate, and Asp44 as a channel lock. These three residues have a fixed position (highly conserved) in influenza A virus M2 channel (Du et al., 2010).
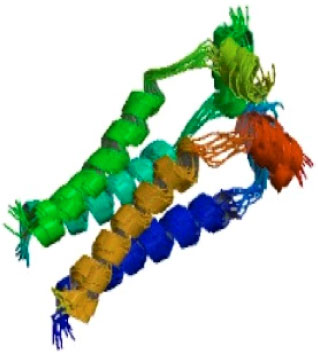 | |
| Fig. 1: | 3D crystal structure of the M2 protein channel, source: RSCB Protein DataBank (http://www.rcsb.org/pdb/explore.do?structureId=2KIH), accessed January 2011 |
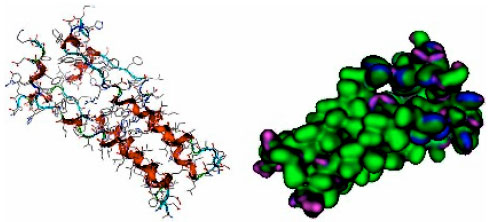 | |
| Fig. 2: | Visualization of M2 channel protein influenza A (H1N1) |
In the tertiary structure of proteins, amino acid residues are located on the hydrophilic exterior (surface), while the hydrophobic residues are generally located on the interior of the protein (Lehninger and Nelson, 2005). Hydrophobicity and hydrogen bond properties of the channel M2 protein of influenza virus A (H1N1) was determined using the software MOE 2008.10. Residues of the M2 channel in Fig. 2 were visualized using the map program in software MOE 2008-10 and utilizing the surface Gaussian contact parameter. The hydrophobic residues are shown in green residues that have H bonds in purple and the polar residues are colored blue.
3D Structure geometry optimization and energy minimization of the M2 channel protein: The process of geometry optimization and energy minimization was done using the software MOE 2008-10. The first stage was carried out with 3D protonation; this was done to alter the enzyme to a protonated state. The application of this 3D protonation was to change the state of the enzyme's ionization level and display the position of hydrogen atoms in the crystal structure. The existence of this hydrogen atom was required in the process of molecular mechanics, dynamics, or the calculation of electrostatic interactions. Then, the potential parameter settings in the setup menu were modified. Parameters to be regulated were related to force field. Selected force field was MMFF94x (Merck Molecular Force Field 94x). It corresponds to peptides, proteins and DNA. MMFF94x was used as a force field because it is considered better than others (Halgren, 1999). Moreover, the sensitivity of the enzyme with a ligand geometry optimization was quite high. MMFF94x can also put hydrogen atoms on the most appropriate position so it is useful also for the validation of the hydrogen atom positions in the solvation state of water molecules. The selected type of gas phase solvation due to the molecular docking stage of the enzyme made in the circumstances would require the removal of rigid solvation energy (Tambunan et al., 2010). The addition of load (partial charge) by using the current method of force field parameters was conducted. The purpose of this addition was to ensure that the proper charge of protonated protein was in the natural state. Henceforth, the docking will run according to its natural state. A fixed hydrogen treatment was used to repair the structure if there was a loss of hydrogen molecule. The next process of protein energy minimization was using MOE 2008-10 software. Energy minimization was done by the RMS gradient of 0.05 kcal Å-1 corresponding to the protein. The purpose of energy minimization was to eliminate undesirable interactions (bad contact) of the structure. Nurbaiti et al. (2010) stated that the initial coordinates of the biomolecules were generally obtained from X-ray crystallography or 3D structure modeling and the distance between each atom could be very close or very far from their equilibrium position. Geometry discrepancies caused the occurrence of undesirable interactions (bad contact) and high-energy steric effects that could result in an unstable simulated system. Moreover, the simulation parameters were adjusted to approximate the real conditions. Then, the minimization process was carried out. It was necessary for restoring the positions of atoms that are not suitable for geometry because the resulting potential energy is too low for the system (Nurbaiti et al., 2010).
Preparation of ligand
Determination of ligands as inhibitors: Determination of protein ligands as inhibitors of the M2 channel was done by modifying the active group of isopinocampheylamine compound. According to Zhao et al. (2011), this compound is an inhibitor for M2 channel protein that can replace amantadine and rimantadine. The mechanism of M2 channel protein inhibitor that is blocking the activity of ion channel protein M2 channel of influenza virus A works by inhibiting viral replication by blocking the flow of protons. The amino group of amantadine and rimantadine, which are crucial as a barrier towards the proton flow, also serve as pharmacopore. Pharmacopore is the group that play a role in the protons’ interaction, whereas, the adamantyl group serves as a framework used to maintain the steric effect. Based on the structure-activities relationship of amantadine, (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine has a structure that may play a role in inhibiting M2 protein channel (Zhao et al., 2011).
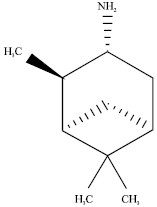 | |
| Fig. 3: | Structure of (1R, 2R, 3R, 5S)-(-)-isopinocampheylamine |
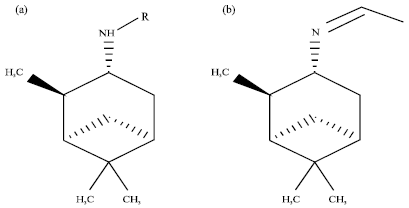 | |
| Fig. 4: | Modification of (1R, 2R, 3R, 5S)-(-)-isopinocampheylamine compound |
Primary amine group in compound (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine could play the same role with the amino group in compound amantadine which is a barrier of proton flow in the influenza virus A. The bicyclic ring of compound (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine also play an equal role with the adamantyl group in amantadine, that is, a framework that provides steric effects (Fig. 3).
To increase the potency of the (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine compound, this study carried out modifications on the existing primary amine group. Modification of (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine was done in two forms. The first form modifies primary amine into secondary amine. Modification of the primary amine into a secondary amine group is expected to increase the activity of these compounds, whereas, the second form is the form of imine. The modified form of the C = N bond is expected to have better interaction with the target protein (Fig. 4).
Active groups that were used to modify the (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine were chosen based on the M2 protein channel inhibitor that was synthesized in an earlier study by Balannik et al. (2009) and Zhao et al. (2011). The active groups are listed in Table 1. This study proved that the active groups were effective for inhibiting M2 proton channel of influenza A H1N1 virus. From the results of modifications of the active groups, 52 ligand candidates for isopinocampheylamine were obtained. A further screening process was carried out to obtain the best three ligand candidates as an inhibitor of the M2 protein channel.
| Table 1: | Structure of active groups |
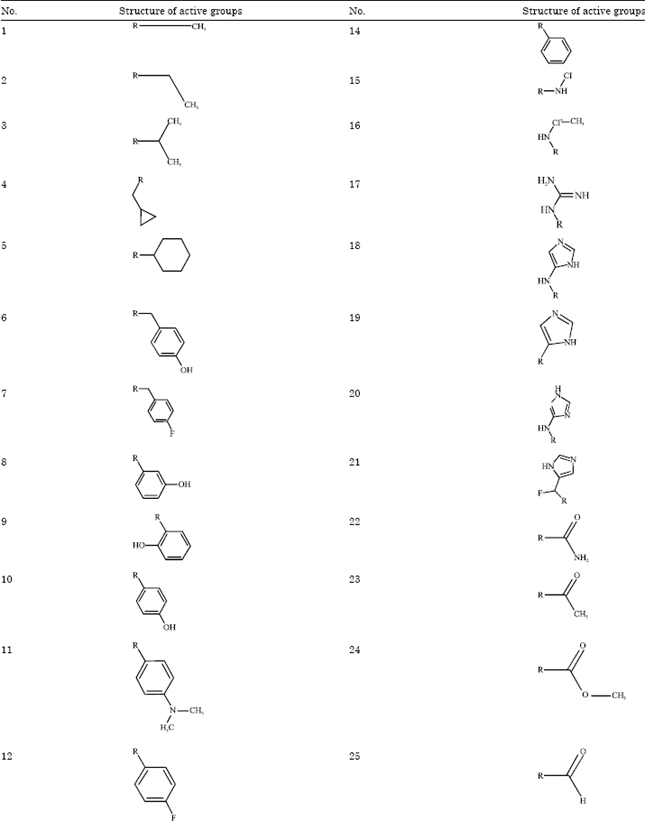  | |
Design of the ligand’s 3D structure: Design of the 3D structure modification of (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine was done using the ACDLabs software. This design was done to prepare the 3D structure modification of isopinocampheylamine compounds as ligands to be used in molecular docking processes. Software ACDlab could describe the position of the active groups of bioactive compounds in accordance with the isolation results.
Storage format of the design modifications was the MDL molfile. Then the format was converted to ligand design using software VEGA MDLmol zz. It means that the software MOE 2008.10 can read the design of these ligands.
Geometry optimization and energy minimization of the ligands' 3D structure: Ligands with MDL mol format were imported into the database viewer (dv) MOE 2008.10 to do geometry optimization and energy minimization. The process of geometry optimization begins with washing of the designed ligands contained in the database viewer MOE 2008-10. The goal is to improve the position of the hydrogen atoms contained in the ligands and also to improve the structure of the ligands. Then, the process was performed using the MMFF94x force field optimization. Partial load settings using the partial charge ligands were conducted with the parameter method of MMFF94x. The parameter method used was appropriate for peptides, proteins and DNA. Then, the energy minimization process was done with the aim of eliminating undesirable interactions. Energy minimization process was performed on the ligands with RMS gradient 0.001 kcal Å-1 (Singh et al., 2007). The purpose of the energy minimization process is to minimize protein interactions in order to eliminate bad contact.
Molecular docking: The M2 channel protein and ligand after optimization were directed to molecular docking processes. The molecular docking process was performed using MOE 2008-10 software. Molecular docking was designed to find the proper conformation bond between the ligand and the receptor. The ligand docking process can form a complex with the enzyme and iterate it to the most optimal conformation (Tambunan et al., 2010). In this study, the docking process was also used for the screening of a number of candidate inhibitors to obtain the best ones that can be bound to the active or binding site of proteins (Teodoro et al., 2001), as well as their potential for development as drugs. Docking process in this study was conducted between 52 ligand candidates and 3 standard ligands (amantadine, rimantadine and isopinocampheylamine). The M2 channel protein functional sites are His37, Trp41 and Asp44. In the process of docking, the protein was pegged to the rigid condition while the ligand was conditioned on the flexible state, in order for free movement and rotation.
The parameters set in the docking process are London dG scoring function. Scoring function is useful in measuring the biological activity based on the bonding and interactions that occur between the ligand and the target protein (Nylander, 2007). Scoring function in MOE 2008.10 is based on force-field scoring and is usually calculated with two energies, that is, the ligand-receptor interaction energy and ligand internal energy (Nylander, 2007). Scoring function is used to retain the London dG (views) of 100, without duplication (Mazur et al., 2010). The “retain” parameter aims to regulate the amount of the best ligand conformation display. London dG shows the magnitude of the Gibbs free energy of binding (ΔGbinding) of each position between the ligand and the enzyme produced by the equation:
where, c is the average rotational and translational entropy gained or released, Eflex is the energy state that reduced the flexibility of the ligand, fHB is the size of the imperfection of the hydrogen bonds’ geometry, CHB is the energy of an ideal hydrogen bond, FM is the size of the imperfections of the geometry of the metal ligations, cM is the energy of an ideal metal ligation and Di is the solvated energy of atom i (MOE tutorials, 2008).
Other parameters were performed in the docking process through software MOE.2008-10 by setting the triangle matcher. Due to the placement of the MOE, the default method was used to demonstrate the random motion of ligand in the enzyme active site to produce an optimal bonding orientation. Triangle matcher was used to orient the ligand in the active site groups based on the charge and spatial fit (Cook et al., 2009). The triangle pose matcher used as much as 1000 which is the default of MOE-Dock program. Stages of refinement were used to make further improvements. Refinements were performed using the force field by the results obtained and it was more accurate when compared with GridMIn using electrostatic calculations on the minimization process (Feher and Williams, 2009). The default setting of refinement forcefield using 6 Å pocket cut off, or the distance of receptors, was included in the docking process (Feher and Williams, 2009). Subsequently, retain (view) parameter of the last refinement results was obtained one by one. Therefore, it was one of the most optimal conformations of each ligand.
Analysis of docking: There are three important things that are generated in docking simulations. The first is the orientation result and position of a ligand as an inhibitor of the enzyme. The second is to identify compounds that have affinity to the protein from the database of available compounds. The third is to predict the affinity of the enzyme molecule that has a targeted simulation docking. These three are scoring functions (London dG) that estimate the value of the ΔGbinding in kcal mol-1. The docking process was carried out 4 times at the screening stage to get the best of the three ligands.
Free energy association (ΔGbinding) and inhibition constants: Tendency of the bond strength may be referred to as the affinity of the ligand to a receptor or enzyme. It can be determined by looking at the value of the ΔG0 binding affinity (kcal mol-1) generated during protein-ligand complexes formation. High affinity of a ligand to the protein is produced from a large intermolecular force between ligands within the protein, while the low affinity of a ligand to the protein is produced from a small intermolecular force between the protein-ligand complexes. When binding free energy values (ΔGbinding) quantified by the KA constant, it is assumed that the biological activity in the conditions of thermodynamic equilibrium is in the formation of protein-ligand complex [EI]. There is a relationship between the values of the bond free energy (ΔGbinding) and those of the inhibitor constants (Ki) which are shown in the following thermodynamic equation (Kitchen et al., 2004):
Based on the formula, the lower or higher the negative value of the ΔGbinding, the stronger the protein-ligand complexes. This is because the stability and strength of non-covalent interactions in protein-ligand complexes can be seen from the large free energy released during the interaction on the protein-ligand complexes formation. In the MOE software, Gibbs free energy (ΔG0) is denoted by S, which shows the total amount of the docking final stage. Value of S has the same score with E_refine, where E_refine is the total energy of the complex bond docking. Docking results showed that from the obtained 52 ligand candidate compounds, there are three best ligands with a negative value ΔGbinding.
From Table 2, there are three ligands (ligands A20, B18 and B20), which have a value of ΔG0 that shows more negativity than that of amantadine, rimantadine and isopinocampheylamine standard. Also from Table 2, the inhibition constant value can also be seen. The dissociation constant of the decomposition herein referred to as Ki may give an idea about the affinity between ligand and decomposition. This constant is reciprocal of the equilibrium constant, thus, Ki is formulated as follows:
where, E is the enzyme, L is the ligand and EL is the enzyme-ligand complex. The smaller the value of Ki, the more the equilibrium reaction tends toward complex formation. Complex protein-ligand binding affinity is said to have a good Ki value if it is in the micromolar scale. Data docking results showed that all ligands have an estimated value of Ki on this scale. Docking data of the software MOE showed inhibition constants in the pKi. This means that the greater the value of the pKi, the smaller the ligand’s Ki. The value of pKi can be used to determine the level of stability in the formation of protein complexes with ligands. From the docking of ligand A20, pKi value is 6.292. The value indicates that A20 has an affinity with the ligand and interacts more strongly with the M2 protein. When compared to other ligands, it forms a complex with the channel better.
| Table 2: | Docking simulation results of binding free energy data |
 | |
| *Standard | |
Hydrogen bonding and contact residues: Apart from the value of Gibbs free energy, or ΔG and inhibition constants, parameters that can be analyzed from the docking process is the interaction between protein and ligand. One of the observed parameters is hydrogen bond interaction. Hydrogen bond is defined as an intermolecular or intramolecular force that occurs between atoms that have a high electronegativity of hydrogen atoms and is covalently bonded to an electronegative atom (Nurbaiti et al., 2010). The criterion for the occurrence of this hydrogen bond is that the distance between the hydrogen with electronegative atoms must be in the range of 2.5-3.5 Å. Hydrogen bonding that occurs in protein-ligand complexes can be identified and analyzed in ligand interaction using software MOE 2008-10.
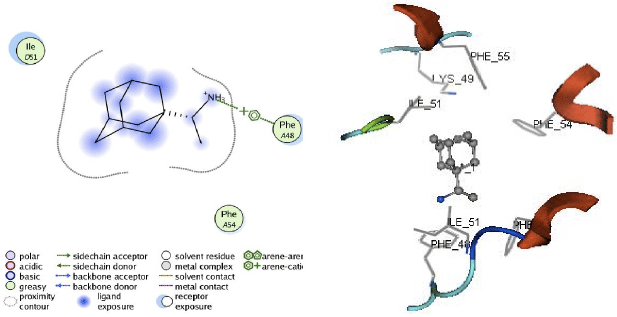
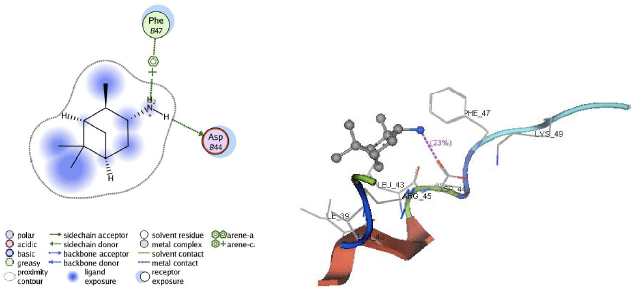
From the analysis of hydrogen bonding in the docking process, it could be seen that the hydrogen bonding interaction between the M2 protein channel and the standard ligands is less than the hydrogen bonding between the M2 protein channel and the three best ligands. In this study, the ligand modification compound of (1R, 2R, 3R, 5S-(-)- isopinocampheylamine showed better activity. From the analysis of hydrogen bonds, it can be seen that these ligands tend to bind to Asp44 which is the target residue. Asp44 residue acts as a channel lock on the M2 protein channel. In normal circumstances, Asp44 residue binds to Trp41 residue to maintain a closed ion channel but at a lower pH, Trp41 residue could be protonated, as such, it weakens the bond between the two ion channels. Thus, it could be opened and may help the virus to release its genetic material. Due to the interaction of the Asp44 residue with ligands, it became more difficult for these residues to be protonated in order to maintain a closed conformation. Aside from being a channel lock, Asp44 residue acts as a proton exit channel on the M2 protein and may also impede the flow of protons. With so many hydrogen bonds on Asp44 residues, it is expected that inhibition on ligand modification function compounds (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine will be better. Table 3 shows that most hydrogen bonding interactions with Asp44 residue occurred in ligand A20. This occurrence was observed on three hydrogen bonds with Asp44 residue in the M2 protein channel (two bonds on chain B and one double bond on chain A). In B18 ligands, two hydrogen bonds occurred with Asp44 residue in the chain of the M2 protein channel D, while the B20 ligands occurred in one hydrogen bond with Asp44 residue chain C. From the results of the standard ligand interaction, it was also seen that amantadine and rimantadine lost their effectiveness. In rimantadine, hydrogen bonding did not occur with the M2 protein channel. Contact residues between rimantadine and the M2 channel protein was not on a functional site. On the interaction between the protein isopinocampheylamine, one hydrogen bond was seen with the functional site residues of Asp44 and it contacted only with the Leu40 residue which is not a functional site on the M2 protein channel.
| Table 3: | M2 protein channels amino acid residue that form hydrogen bonds with the ligand |
 | |
| *Standard; residue in bold font are the functional site residue of M2 channel protein | |
Apart from hydrogen bonding interactions, the analysis of docking results can also be viewed from the contact between the protein residues and the ligand. Interaction of non-covalent or non-bonding occurring between the protein and ligand can increase the ligand affinity to the protein. Non-bonding interactions represent a flexible interaction between pairs of atoms and particles. Two types of non-bonding interactions that can lead to the most common change in potential energy are the electrostatic interaction and van der Waals interactions. Therefore, there should be an analysis of the docking simulation results of the contact residues’ protein-ligand complexes, such that the type of enzyme residue that interacts with the ligand can be known. The analysis shows that the contact residues of the ligand modification of compound (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine are better than those of the standard ligand. In addition to the functional site of interaction with Asp44, ligand modification of compound (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine also has a contact residue with another functional site. This is Trp41 residue in ligands A20 and B20. This is expected to increase the effectiveness of the M2 protein inhibitors of this channel and it will be displayed following the 2D visualization and 3D complex interactions of the best candidate ligand and standard ligand with M2 protein channel. 2D visualization is shown on the left while 3D visualization is shown on the right. In 3D visualization, hydrogen bonds between ligands with proteins are marked with purple colored dashed lines (Fig. 5-10).
Drug scan analysis: The three best ligand docking properties analysis was tested for their similarities with an existing drug (drug likeness) using the Lipinski rules (Lipinski's Rule of Five). Lipinski's Rule of Five helps to distinguish the difference between drug-like and non drug-like molecules by taking into account the extent of absorption or permeability of the lipid bilayer present in the human body. The rule predicts that the drug likeness probability is high enough if it has two or more criteria as given thus:
| • | A molecular weight of less than 500 mg mol-1 |
| • | A high lipophilicity (logP less than 5) |
| • | Hydrogen bond donors less than 5 |
| • | Hydrogen bond acceptor less than 10 |
| • | Refractory molarity between 40 and 130 (optional) (SCFBIO-IITD) |
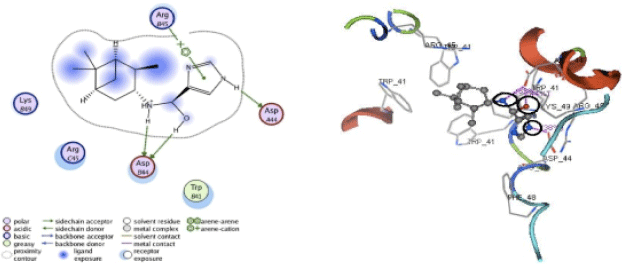 | |
| Fig. 5: | Visualization of 2D and 3D interaction of A20 ligand with M2 channel protein |
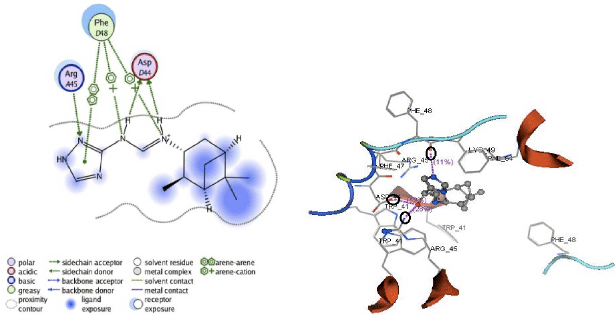 | |
| Fig. 6: | Visualization of 2D and 3D interaction of B18 ligand with M2 channel protein |
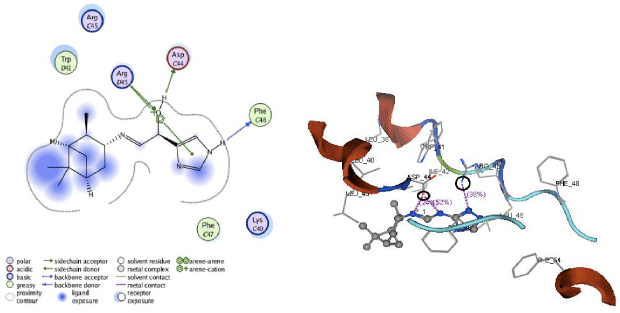 | |
| Fig. 7: | Visualization of 2D and 3D interaction of B20 ligand with M2 channel protein |
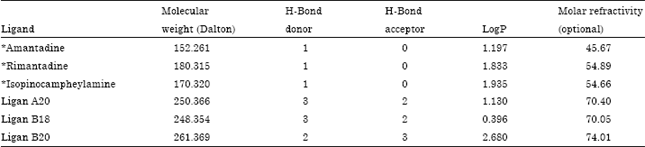
Table 4 shows the result of drug likeness based on ligand screening using Lipinski's online software filter (SCFBIO-IITD). From Table 4, it could be seen that the best ligand candidates’ molecular weight is less than 500 mg mol-1, which fulfills the criteria of Lipinsky's Rule. The value of the hydrogen bond donor and acceptor was also less than the maximum extent permitted by Lipinsky's Rule. LogP values of the three best ligands or ligand candidates also met the standards of Lipinsky's Rule. LogP value is the partition coefficient that is defined as the ratio of the concentration of a molecule in octanol and water.
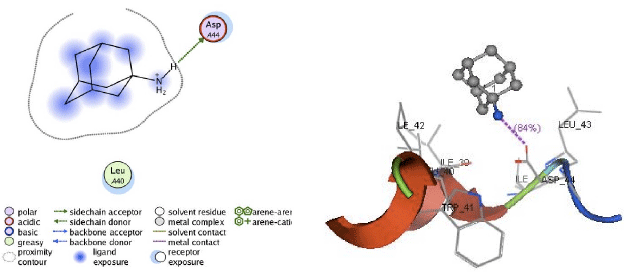 | |
| Fig. 8: | Visualization of 2D and 3D interaction of amantadine with M2 channel protein |
 | |
| Fig. 9: | Visualization of 2D and 3D interaction of rimantadine with M2 channel protein |
| Table 4: | Drug scan result of the best three ligands and standard ligands |
 | |
| *Standard | |
 | |
| Fig. 10: | Visualization of 2D and 3D interaction of isopinocampheylamine with M2 channel protein |
Analysis of drug scans was performed on the two best ligand dockings which has the lowest scoring function. LogP value is related to the hydrophobicity of drug molecules. The larger the logP value, the more hydrophobic the molecule. Drugs molecule that are too hydrophobic tend to have greater toxicity due to their ability to be held longer in the lipid bilayer and distributed more widely in the body. This would cause reduction of the selectivity of binding to the target enzyme. LogP values that are too negative cannot be recommended because if the compounds are too hydrophilic, it could not pass through the lipid bilayer and it is possible to interact with water solvent. The last criterion is the value of the molar refractivity. According to Lipinsky's Rule of molar refractivity, the values that were allowed were between 40 and 130. The three candidates met the rules of the ligand as well as the standard ones. Molar refractivity is a measure of the total polarizability of a drug molecule which is very dependent on temperature, refractive index and pressure. Determination of the molar refractivity can be done using the Lorentz-Lorenz formula:
where, M is the molecular weight, η is the refractive index and r is the density whose value depends only on light waves that are used to measure the refractive index. From the analysis of drug-scan using Lipinsky's Rule of Five, it could be concluded that the three best candidates were possible candidates for oral drugs.
In addition to Lipinsky's Rule of Five, the value of drug likeness can also be seen from the level of oral bioavailability. High oral bioavailability often becomes an important consideration for developing bioactive molecules as therapeutic agents (Veber et al., 2002; Tambunan and Wulandari, 2010). Oral bioavailability is the extent to which a drug or other materials are available to the target tissue after administration of a drug or substance (Tambunan and Wulandari, 2010).
| Table 5: | Oral bioavailability screening results of standards and ligands (Egan’s rules) |
 | |
| *Standard | |
Screening level of ligand's oral bioavailability could be done using Egan's rules (Egan et al., 2000). Based on Egan's Rules, a molecule is said to have a good level of oral bioavailability if it meets the following criteria:
First, the screening was done by determining the ligand properties by using the online software molinspirations (molinspirations.com). The results of the oral bioavailability screening of the standards and ligands are shown in Table 5.
From the results of oral bioavailability screening of standards and the ligands using Egan's Rules, it is seen that the three best candidates met the criteria of Egan's Rules. It could be concluded that the three best candidates are ligands A20, B18 and B20 which have high levels of good oral bioavailability.
Inhibitor toxicity prediction nature: Testing the toxicity properties of the three-channel M2 protein inhibitor candidates obtained from the previous process is important as the basis for drug determination. It is necessary to predict their adverse effects to living species. One method that can be used in toxicological studies is qualitative or quantitative structure-activity relationship (QSARs). The results of studies conducted using animals are the basis of QSARs in toxicological studies, because it could not guarantee the completeness of the data required for the toxicological studies. These studies have some limitations such as funds, requires too much time, availability of adequate laboratory and the issues about the ethics of using animals as test materials (Gonzalez-Diaz et al., 2009). To help overcome these problems, several tools have been developed which can perform quick and inexpensive toxicological studies or determination of the toxicological properties of a molecule. Some software developed to assist researchers in determining the toxicological properties of a molecule are: ToxTree (Ideaconsult Ltd., Bulgaria) and Lazar (http://lazar.in-silico.de/).
Both software are used in determining the toxicological properties of the three-ligand results of previous screening of ligands A20, B18 and B20. Determinations of the toxicological properties of the three ligands are focused on carcinogenicity and mutagenicity because they are an important concern in human health and deal directly with the aim of the drug design.
The difference between the two software is the basis for determining the toxicological properties of a molecule. The Toxtree is based on rules of Benigni/Bossa for mutagenicity and carcinogenicity, and was developed by Romualdo Benigni and Cecilia Bossa from the Instituto Superiore in Sanita, Rome, Italy and approved by the European Chemical Bureau, Institute for Health and Consumers Protection, European Commission-Joint Research Centre (JRC) in 2008. Some things to consider in determining the toxicological properties using Toxtree are the presence or absence of genotoxic and nongenotoxic structural alerts (SAS) and QSARs determination.
| Table 6: | Toxtree toxicity prediction results |
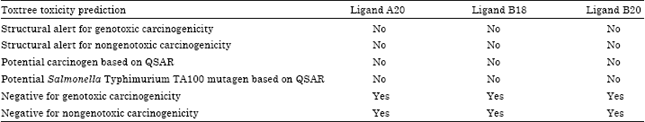 | |
| Table 7: | Results of Lazar toxicity prediction |
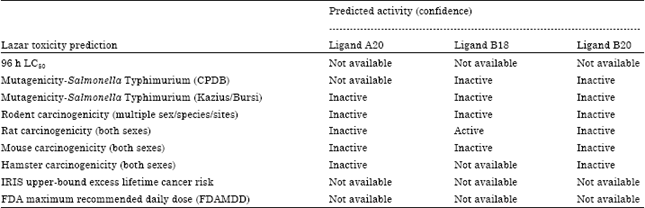 | |
Benigni and Bossa rules are based on the existence of groups which are potentially seen as having mutagenic and carcinogenic properties of the test compound. Table 6 shows the result of the determination of the toxicological properties of the software Toxtree.
From the results of the toxicological prediction using software Toxtree, it could be seen that the three ligands have no structural alerts (SAS), which are genotoxic and nongenotoxic and also with this approach, QSARs which are neither mutagenic nor carcinogenic, were not found. As for the software online Lazar, predictions using this software were based on structural equation fragments of a molecule compared with the structural fragment toxicology that exist in databases of known carcinogenicity and mutagenicity and the test results using test animals and microbes. Table 7 shows the result of the determination of the toxicological properties of software Lazar.
The results of software Lazar could predict mutagenic properties of the three ligands in Salmonella Typhimurium by two methods of CPDB and Kazius/Bursi. The three ligands of the test showed negative results. The carcinogen properties were predicted by testing them against animals such as rodents (rodent), rat (rat), murine (mouse) and hamster. The test results using all three animals showed negative carcinogenicity properties; only the B18 ligands showed positive carcinogenic results which could be active against rats. Some parameters in software Lazar did not show any result (not available) for the three ligands. This was caused by the less availability of data in the structural fragment and the inadequate support to predict the outcome of those parameters such as LC50 and the daily dose.
CONCLUSION
This study obtained 52 ligand modifications for compound (1R, 2R, 3R, 5S)-(-)-isopinocampheylamine. In the process of docking, three of the best ligands were obtained. They have the lowest value of ΔGbinding and the best interaction with the M2 protein channel compared with the standard ligand of amantadine, rimantadine and isopinocampheylamine. The three ligands are A20, B18 and B20. From the obtained results by docking the data, the three ligands form hydrogen bonds with a residual functional M2 channel, that is, Asp44. Analysis of the drug-scan gives good results for the three best ligands based on Lipinski’s rules. Toxicological properties of ligands A20, B18 and 820 as a whole are predicted not to be carcinogens and mutagens.
However, further steps like the molecular dynamic simulation to determine the effect of temperature and solvent on ligand interaction with the M2 protein channel and ADME analysis and bioactivity of the ligand which has been designed to determine the treatment of human body systems of ligands, need to be considered.
ACKNOWLEDGMENTS
This research was supported by Hibah Pasca, DIKTI, Indonesia Ministry of Education. The authors are thankful to DRPM, University of Indonesia for facilitating this research. U.S.F.T was supervising this research, R. H was working on the technical details and A.A. P was preparing the English manuscript and re-verifying the data.
REFERENCES
- Alsaif, A., O. Al-Sagair, A. Albarrak, I.A.M. Ginawi, T.D. Hussein and M.E. Sweelam, 2010. Hail community acceptance of A/H1N1 vaccine. J. Med. Sci., 10: 162-168.
CrossRef - Amir, A., M.A. Siddiqui, N. Kapoor, A. Arya and H. Kumar, 2011. In silico molecular docking of influenza virus (PB2) protein to check the drug efficacy. Trends Bioinform., 4: 47-55.
CrossRefDirect Link - Balannik, V., J. Wang, Y. Ohigashi, X. Jing and E. Magavern et al., 2009. Design and pharmacological characterization of inhibitors of amantadine: Resistant mutants of th M2 ion channel of influenza a virus. Biochemistry, 48: 11872-11882.
Direct Link - Beigel, J.H., J. Farrar, A.M. Han, F.G. Hayden and R. Hyer et al., 2005. Avian influenza A (H5N1) infection in humans. N. Engl. J. Med., 353: 1374-1385.
CrossRefPubMedDirect Link - Betakova, T., 2007. M2 Protein-A proton channel of influenza a virus. Curr. Pharm. Des., 13: 3231-3235.
Direct Link - Cheng, P.K.C., T.W.C. Leung, E.C.M. Ho, P.C.K. Leung, A.Y.Y. Ng, M.Y.Y. Lai and W.W. Lim, 2009. Oseltamivir- and amantadine-resistant influenza viruses A (H1N1). Emerg. Infect. Dis., 15: 966-968.
Direct Link - Cook, I.T., T.S. Leyh, S.A. Kadlubar and C.N. Falany, 2009. Structural rearrangement of SULT2A1: Effect sondehydroepiandrosterone and raloxifene sulfation. Horm. Mol. Biol. Clin. Invest., 1: 81-87.
PubMedDirect Link - De Clercq, E., 2006. Antiviral agents active against influenza A virus. Nat. Rev. Drug Discov., 5: 1015-1025.
Direct Link - De Jong, M.D., T.T. Thanh, T.H. Khanh, V.M. Hien and G.J.D. Smith et al., 2005. Oseltamivir resistence during treatment of influenza A (H5N1) infection. N. Engl. J. Med., 353: 2667-2672.
Direct Link - Du, Q.S., R.B. Huang, S.Q. Wang and K.C. Chou, 2010. Designing inhibitors of M2 protein channel against H1N1 swine influenza virus. PLoS one, Vol. 5.
CrossRef - Egan, W.J., K.M. Jr. Merz and J.J. Baldwin, 2000. Prediction of drug absorption using multivariate statistic. J. Med. Chem., 43: 3867-3877.
CrossRefDirect Link - Feher, M. and C.I. Williams, 2009. Effect of input differences on the results of docking calculations. J. Chem. Inform. Model., 49: 1704-1714.
CrossRef - Gonzalez-Diaz, H., F. Prado-Prado, L.G. Perez-Montoto, A. Duardo-Sanchez and A. Lopez-Diaz, 2009. QSAR models for proteins of parasitic organisms, plants and human guests: Theory, applications, legal protection, taxes and regulatory issues. Curr. Proteomics, 6: 214-227.
CrossRef - Halgren, T.A., 1999. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem., 20: 720-729.
CrossRef - Hu, W., S. Zeng, C. Li, Y. Jie, Z. Li and L. Chen, 2010. Identification of hits as matrix-2 protein inhibitors through the focused screening of a small primary amine library. J. Med. Chem., 53: 3831-3834.
CrossRef - Karim, S.A.A. and R. Razali, 2011. A proposed mathematical model of influenza A, H1N1 for Malaysia. J. Applied Sci., 11: 1457-1460.
CrossRefDirect Link - Kitchen, D.B., H. Decornez, J.R. Furr and J. Bajorath, 2004. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discovery, 3: 935-949.
CrossRefDirect Link - Lew, W., X. Chen and C.U. Kim, 2000. Discovery and development of GS 4104 (oseltamivir) an orally active influenza neuraminidase inhibitor. Curr. Med. Chem., 7: 663-672.
Direct Link - Mazur, P., T. Magdziarz, A. Bak, Z. Chilmonczyk and T. Kasprzycka-Guttman et al., 2010. Does molecular docking reveal alternative chemopreventive mechanism of activation of oxidoreductase by sulforaphane isothiocyanates?. J. Mol. Model., 16: 1205-1212.
CrossRef - Nurbaiti, S., H. Nagao, H. Saito, R. Hertadi, M.A. Martoprawiro and Akhmaloka, 2010. Domain motions of Klenow-like DNA polymerase I ITB-1 in the absence of substrate. Int. J. Integr. Biol., 9: 104-110.
Direct Link - Lakshmi, P.T.V., S. Radhika and A. Annamalai, 2011. Molecular docking analysis of phyto-ligands with multi drug resistant β-lactamases of Staphylococcus aureus. Trends Bioinform., 4: 23-34.
CrossRefDirect Link - Rungrotmongkol, T., P. Intharathep, M. Malaisree, N. Nunthaboot and N. Kaiyawet et al., 2009. Susceptibility of antiviral drugs against 2009 influenza A (H1N1) virus. Biochem. Biophys. Res. Commun., 385: 390-394.
CrossRef - Sar, T.T., P.T. Aernan and R.S. Houmsou, 2010. H1N1 influenza epidemic: Public health implications for Nigeria. Int. J. Virol., 6: 1-6.
CrossRefDirect Link - Singh, H., S.S. Marla and M. Agarwal, 2007. Docking studies of Tau protein. Int. J. Comput. Sci., 33: 1-7.
Direct Link - Sohail, M.N., F. Rasul, A. Karim, U. Kanwal and I.H. Attitalla, 2011. Plant as a source of natural antiviral agents. Asian J. Anim. Vet. Adv., 6: 1125-1152.
CrossRef - Sur, S., G. Sen, S. Thakur, A.K. Bothra and A. Sen, 2009. In silico analysis of evolution in swine flu viral genomes through re-assortment by promulgation and mutation. Biotechnology, 8: 434-441.
CrossRefDirect Link - Tambunan, U.S.F., Fadilah and A.A. Parikesit, 2010. Bioactive compounds screening from zingiberaceae family as influenza a/swine flu virus neuraminidase inhibitor through docking approach. Online J. Biol. Sci., 10: 151-156.
Direct Link - Tambunan, U.S.F. and E.K. Wulandari, 2010. Identification of a better Homo sapiens class II HDAC inhibitor through binding energy calculations and descriptor analysis. Bio Med. Cent. Bioinform., Vol. 11.
CrossRef - Tambunan, U.S.F., O. Hikmawan and T.A. Tockary, 2008. In silico mutation study of haemagglutinin and neuraminidase on banten province strain influenza a H5N1 virus. Trends Bioinform., 1: 18-24.
CrossRefDirect Link - Webby, R.J. and R.G. Webster, 2001. Emergence of influenza A viruses. Philos. Trans. R. Soc. Lond B Biol. Sci., 356: 1817-1828.
CrossRef - Veber, D.F., S.R. Johnson, H.Y. Cheng, B.R. Smith, K.W. Ward and K.D. Kopple, 2002. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem., 45: 2615-2623.
CrossRefDirect Link - Yahyapour ,Y., R. Hamkar, A. Moradi, M. Mahmoudi, Z. Nourozbabaei, Z. Saadatmand and T.M. Azad, 2007. Isolation and typing of the influenza viruses in the caspian littoral of Iran. J. Med. Sci., 7: 307-310.
CrossRef - Zhao, X., C. Li, S. Zeng and W. Hu, 2011. Discovery of highly potent agents againts influenza A virus. Eur. J. Med. Chem., 46: 52-57.
CrossRef