
A. Falodun
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Benin, Benin City, Nigeria
I.M. Qadir
H.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi 75270, Pakistan
C.F. Poh
Department of Pharmacology, Faculty of Basic Medical Sciences, College of Health Sciences, Niger Delta University, Wilberforce Island, Yenagoa, Nigeria
K.I. Omogbai Eric
Department of Pharmacology and Toxicology, Faculty of Pharmacy, University of Benin, Benin City, Nigeria
M.I. Choudhary
H.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi 75270, Pakistan
Research Journal of Phytochemistry
Year: 2009 | Volume: 3 | Issue: 2 | Page No.: 35-43







ABSTRACT
One iridoid and 2 phenylpropanoid glycosides were isolated from the stem bark of S. kunthianum together with mixtures of β-sitosterol and β-sitosterol glucoside. Their structures were determined by IR, HRESIMS and 1D and 2D NMR experiments and their enzyme inhibitory effect evaluated using xanthine oxidase. The inhibitory activities of 6-O-trans-p-coumaroyl-decinnamoylglobularimin-Stereospermiside, (3, 4-dihydroxyphenyl) -ethyl-O- α-rhamnopyranosyl (1→ 3) -4-O cinnamoyl - β-D glucopyranoside and 1,6 di-O-cinnamoyl-β-D-glucopyranoside 1-3 were evaluated and compared to the standard positive control.
PDF Abstract XML References Citation
How to cite this article
A. Falodun, I.M. Qadir, C.F. Poh, K.I. Omogbai Eric and M.I. Choudhary, 2009. Bioactive Chemical Constituents of Stereospermum kunthianum (Bignoniaceae). Research Journal of Phytochemistry, 3: 35-43.
URL: https://scialert.net/abstract/?doi=rjphyto.2009.35.43
URL: https://scialert.net/abstract/?doi=rjphyto.2009.35.43
INTRODUCTION
Stereospermum kurthianum (Charm, Sandrine Petit), family Bignoniaceae is a medicinal plant widely used by the people of Sudano-Guinea savanna as remedy against cough, rheumatism, ulcers, leprosy and respiratory infections (Vonmaydell, 1986). Extensive information about morphological, karyological and ecogeographic differentiation of the genus has been accumulated (Gill, 1992). Decoctions of the stem bark of S. kunthianum is known to possess antiseptic and diuretic properties. Xanthine is a key enzyme that catalyzes the oxidation of hypoxanthine to xanthine and then xanthine in the presence of molecular oxygen as electron acceptor, to yield uric acid, superoxide anions and hydrogen peroxide (Constantino et al., 1998). The inhibition of this enzyme is thereful useful for the treatment of diseases such as kidney stone, gout and inflammatory conditions.
In continuation of the chemotaxonomic and pharmacological investigation on the genus Stereospermum (Ching et al., 2008) herein, we report the isolation and xanthine oxidase inhibitory effect of the plant secondary metabolites.
MATERIALS AND METHODS
Silica gel (70-230 mesh) and sephadex LH-20 were used for the column chromatography. Thin layer chromatography was performed on pre-coated silica gel G-25-UV254 plates. Detection was carried out at 254 and 366 nm by ceric sulphate. For recycling HPLC (LH 908W), a semi preparative (ODS-H80) was used. Optical rotations were measured on a Jasco-DIP-360 digital polarimeter. The UV and IR spectra were recorded on Hitachi-UV-3200 and Jasco-320-A spectrophotometer, respectively. NMR spectra were run on Bruker Avance-500 spectrometers EIMS, ESI and FABMS spectra were recorded on a JMS-HX-110 spectrometer.
Plant Material
The fresh stem bark of S. kunthianum was collected in March, 2006 in Ogun State Nigeria. Botanical identification and authentification was done by Mr. Usang Felix of the Forest Research Institute of Nigeria, Ibadan. A voucher specimen (No. FHI 107277) was deposited in the same institute (FRIN).
Extraction and Isolation
The shade dried stem bark (500 g) was exhaustively extracted with MeOH (4x5 Lx48 h) at room temperature. The extract was evaporated to dryness to yield a residue (80 g), which was dissolved in water (2.5 L) and partitioned with hexane, chloroform, ethyl acate and n-butanol. The n-butanol soluble fraction (30 g) was subjected to vacuum liquid chromatography over silica gel using CHCl3-MeOH, gradient upto 100% methanol to obtain 20 fractions (1-20). Fraction 17 was subjected to flash silica gel chromatography using MeOH:CHCl3 (30:70) and (40 : 60) followed by column chromatography over Sephadex LH-20 with distilled water to get semi pure compounds 1-3 which were finally purified on recycling preparative HPLC (L - H80 preparative column, MeOH : H2O (1:1), flow rate (4 mL min-1), detection (UV and RI detectors), tR 12 min (1, 8.6 mg), 25 min (2, 11.3 mg) and 36 min (3, 9.8 mg).
Identification of Sugar
The sugar attached to the aglycone part were hydrolyzed by 2N HCl, identified as glucose in 1, glucose and rhamnose in 2 and glucose in 3, on silica gelco-TLC with standards (Sigma-Aldrich) (BAW 4.1:5).
Xanthine-Xanthine Oxidase Activity
Absorbance was measured on a spectramax 340 microplate reader (Molecular Devices). Xanthine (X-0626) and xanthine oxidase (EC 1.1.3.22) (from butter milk) (Sigma Aldrich, Japan) were used. Xanthine oxidase activity was carried out by a modified method of Candan (2003). The reaction mixture containing 10 μL of 1 mM pure sample was dissolved in DMSO, 125 μL of Phosphate buffer (0.1 M, pH 7.4), 0.003 units of xanthine oxidase dissolved in buffer (25 μL) and 25 μL of 0.15 mM xanthine as substrate for enzyme. After addition of xanthine oxidase, the mixture was incubated for 10 min and pre-read in the UV region (λmax 295). The substrate was added to reaction mixture and final continuous reading for 30 min at an interval of 1 min was observed. The percentage inhibitory activity of the compounds were determined against a DMSO blank and calculated using the following formula:
of samples was determined by using EZ-FIT windows-based software.
Statistical Analysis
Data obtained were expressed as Mean±SEM. Statistical analysis done by one way Analysis of Variance (ANOVA) and student’s t-test. SARS statistical package was used in the analysis of data.
RESULTS AND DISCUSSION
The methanol extract of the stem bark of Stereospermum kunthianum was partitioned between n-hexane, chloroform, ethylacetate and n-butanol. The n butanol soluble part, upon repeated Column Chromatography (CC) over silica gel and High Pressure Liquid Chromatography (HPLC) afforded 3 pure compounds. By means of spectroscopic and published information they were identified as iridoid ester glycoside compound (1) and two phenylpropanoid glucosides compounds (2 and 3).
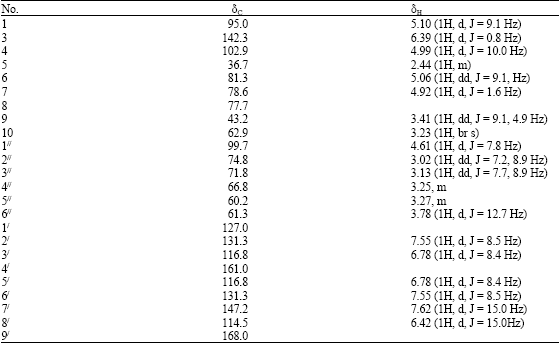
The molecular formula of compound 1 was determined as C24H30O13 by its FAB-ESIMS analysis to be 526.4872. The 1H NMR spectrum demonstrated the presence of 4 aromatic signals assigned to the phenyl group δ 7.55 (1H, d, J = 8.5 Hz), 6.78 (1H, d, J = 8.4 Hz), 6.78 (1H, d, J = 8.4 Hz), 7.55 (1H, d, J = 8.5 Hz), for H-2/, H-3/, H-5/ and H-6/, respectively, while the remaining protons at δ 5.10 (1H, d, J = 9.1 Hz), 6.39 (1H, d, J = 6.7 Hz), 4.99 (1H, d, J = 10.0 Hz), 2.44 (1H, m), 5.06 (1H, dd, J = 9.1 Hz), 4.92 (1H, d, J = 5.1 Hz), 3.41 (1H, dd, J = 69.1, 4.9 Hz) for H-1, H-3, H-4, H-5, H-6 and H-7 and H-9, respectively. There were conspicuous presence of AB type of methylene protons at δ 3.20 and 3.16 (each d = 10.8 Hz), assigned to H-10. The assignment of the phenylpropanoid moiety as trans was due to the presence of the coupling constant of the proton signals at δH 6.42 and 7.62 (each, J = 15.0 Hz). The structure agreed with previously reported data of an irridoid glycoside (Tripetch et al., 2006).
The 13C NMR spectrum summarized in Table 1 was assigned on the basis of Broad Band (BB), Distortionless Enhancement Polarization Transfer (DEPT), Heteronuclear Multiple Quantum Coherence (HMQC) experiments. The appearance of the anomeric carbon resonating at δ 99.7 suggested the presence of one monosaccharide moiety. The 13C NMR spectrum also revealed 24 carbon atoms in the molecule, 6 carbon signals were seen for the sugar moiety, confirming the presence of 1 hexose sugar, 9 carbon signals for the phenylpropanoid moiety, the remaining assignable to the aglycone moiety.
| Table 1: | 13C NMR (500 MHZ) and 1HNMR (500 MHZ) chemical shifts of 1 |
 | |
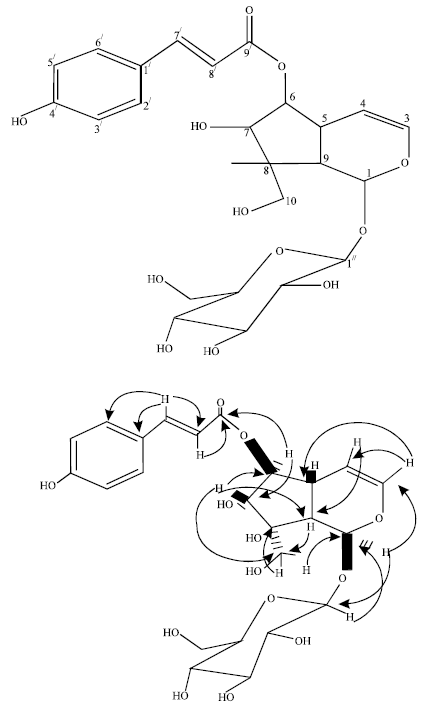 | |
| Fig. 1: | Selected HMBC correlations of 1 |
The analysis of the DEPT experiments indicated that compound 1 (Fig. 1) contained several methines and 1 methylene (δ 62.9) for the aglycone part, consistent with an iridoid skeleton (Chaudhuri and Sticher, 1981).
The 13C NMR data analyzed were similar to the reported data of Stereospermum cylindricum (Tripetch et al., 2006). The chemical shift of δ 95.0 assignable to the acetal moiety in C-1 was conspicuously evident in the spectral. And on the basis of all the above evidences the structure of 1 was established as 6-O-trans-p-coumaroyl-decinnamoylglobularimin known as Stereospermiside. However, this is the first report of this irridoid glycoside from the stem bark of Stereospermum kunthianum. The acid analysis of compound 1 yielded the aglycone and the sugar. The sugar obtained from hydrolysate was identified as glucose by TLC comparison with standard sample and the J values data deduced the β-D-pyranosyl configuration for configuration of glucose molecule (Lanzetta et al., 1984). There is a wide distribution of iridoids in plants belonging to Bignoniaceae family (Lino von Poser et al., 2000) and are well known for their broad spectrum of biological activity (Sticher, 1997). Thus, the chemical ecology of the plant could be useful in further research in Bignoniaceae.
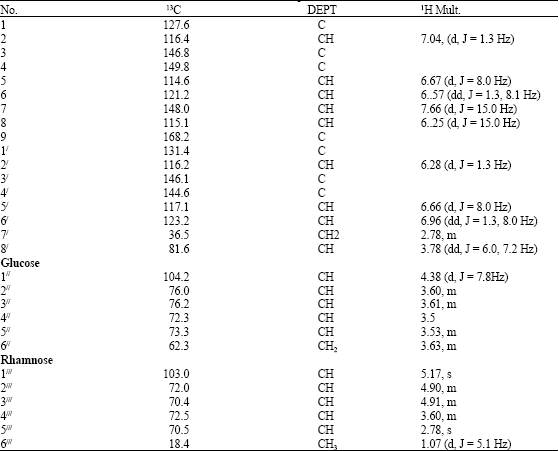
Compound 2 gave m/z 624.5875 for C29H36O15 by EI-ES/IMS. Bands for hydroxyl (3418 cm-1) and ester carbonyl (1032 cm-1) functional groups were suggested by IR spectroscopy and the UV absorption at 234 and 254 nm was typical of cinnamic acid ester derivative as reported by (Lildihone et al., 2005). The 1H NMR spectra (Table 2) displayed signals attributable to the cinnamoyl and 3, 4- dihydroxy phenyl groups with two conspicuous ABX systems at δ 7.04 (d, J = 1.3 Hz), 6.67 (d, J = 8.0 Hz), 6.57 (dd, J = 1.3, 8.1 Hz) and δ 6.28 (d, J = 1.3 Hz), 6.60 (d, J = 8.0 Hz) and 6.96 (dd, J = 1.3, 8.0 Hz) assignable to δ 116.4 (C-2), 114.6 (C-5), 121.2 (C-6) and 116.2 (C-2/), 117.6 (C-5/), 123.2 (C-6/), respectively. Additionally, the signals at δ 2.78 (m) and 3.78 (dd, J = 6.0, 7.2 Hz) could be ascribed to benzylic oxymethylene protons, thus revealing the second aromatic portion of 2 as a benzylic system. The signals at δ 6.25 (d, J = 15.0 Hz) and 7.66 (d, J = 15.0 Hz) could be attributed to trans olifinic protons characteristic of a cinnamoyl moiety. Also, observed were the anomeric protons of O-β- D-glucosyl and rhamnose moieties at δ 4.38 (d, J = 7.8 Hz) and 5.17 (s), respectively.
| Table 2: | 1H NMR (500 MHZ) and 13C NMR (500 MHZ) of compound 2 |
 | |
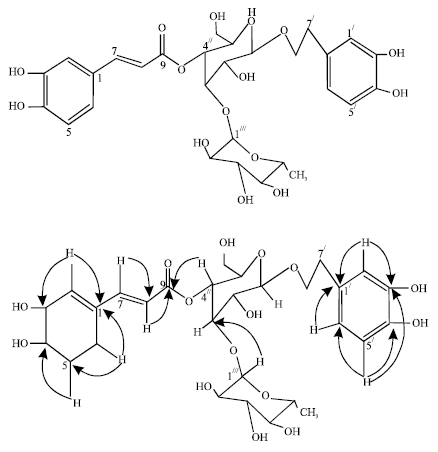 | |
| Fig. 2: | Selected HMBC correlations of 2 |
The 13C NMR (Table 2) displayed 29 carbon signals of which 12 were assignable to the 2 sugar (glucose and rhamnose), 9 to the phenylpropanoid ester, while the remaining were ascribed to the benzylic system. Two anomeric methine carbons at δ 104.2 and 103.0, several hydroxymethines, one hydroxymethelene at δ 62.3 and 1 methyl group at δ 18.4 resembles to those of glucose and rhamnose respectively and unequivocally suggested that Fig. 2 possessed glucopyranosyl and rhamnopyranosyl units. The 1.7 ppm up-field shift of C-4 in the 13C NMR spectrum indicated the attachment of cinnamic acid group to position (C- 4//) of the glucose.
The HMBC correlations between H - 4// (δ 3.50, m) and the carbonyl (δ 168.2, C-9) proved the β-glycosidic linkage of glucose to the carbonyl of the cinnamic acid moiety. HMBC correlations (Fig. 3) between H-1/// of rhamnose and glucosyl C-3// confirmed the linkage point of the sugar moiety and the structure was then determined as (3, 4-dihydroxyphenyl)-ethyl-O-α-rhamnopyranosyl (1→3)-4-O cinnamoyl-β-D glucopyranoside.
Compound 3 gave m/z 504.1268 for C24H24O12 by FAB MS analysis. Acid hydrolysis of 3 gave D-glucose as identified by Co-TLC and GC-MS. 1HNMR signals (Table 3) were evident for a doubled trans cinnamoyl moiety with olefinic protons at δ 7.67 and 6.28 coupled by 15.08 Hz and another one at δ 7.57 and 6.31 coupled by 15.9 Hz revealing the presence of 2 trans olifinic protons in the molecule. The 1H NMR of compound 3 also displayed aromatic protons of ABX systems. The presence of a β-D glucosyl moiety at δ 5.57 (d, J = 7.7 Hz) indicating a β orientation of the glucose was clearly shown in the spectrum.
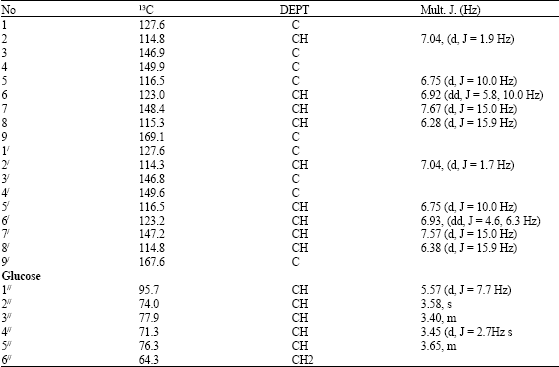
| Table 3: | 1HNMR (CD3OD, 500Hz) 13C NMR (CD3OD, 500 MHZ) of compound 3 |
 | |
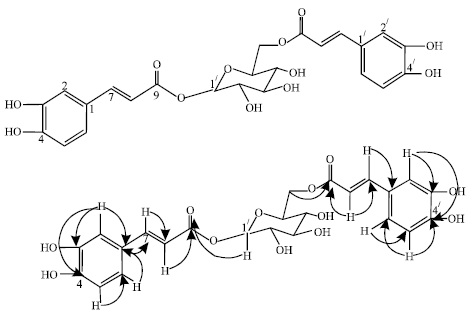 | |
| Fig. 3: | Important HMBC correlations of 3 |
The HMQC NMR correlation between H-1// (δ 5.57) and C-9 (δ 169.0) proved the β-glycosidic ester linkage with cinnamic acid. The 1H and 13C NMR data of 3 was similar to garashangin (Hiroko et al., 1988). The 13C NMR of compound 3 displayed 24 carbon signals, 18 were assignable to the 2 molecules of phenylpropanoid esters while the remaining 6 accounted for the 1 hexose unit. The linkage of sugar and the aglycone was determined mainly by HMBC experiment (Fig. 3). Complete assignment of the protons in each sugar system was achieved by considering the 1D and 2D TOCSY, 1D and 2D NOESY and the 1H-1H COSY spectra, while the carbons were assigned from HMQC and HMBC data. A 1D TOCSY experiment obtained by irradiating at the well resolved anomeric protons of glucose at δ 5.57 (1H, d, J = 7.7 Hz) a set of coupled oxymethine protons of the sugar and an oxymethylene at δ 64.3.The structure was then determined as 1,6 di-O-cinnamoyl-β-D-glucopyranoside called Stereostin and the first time to be reported in this plant.
| Table 4: | In vitro inhibition of xanthine by 1-3 |
 | |
| SEM: Standard error of mean | |
The acid hydrolysis of compounds 1-3 with 2N HCl gave their respective aglycones and glucose and rhamnose as the sugar on co-TLC with standard samples.
In the search for natural products with xanthine oxidase inhibitory activity, Stereospermiside, (3, 4-dihydroxyphenyl)-ethyl-O-α-rhamnopyranosyl (1→3)-4-O cinnamoyl -β-D glucopyranoside and 1,6 di-O-cinnamoyl-β-D-glucopyranoside (1→3) were tested for in vitro inhibitory activities against this enzyme. Their IC50 values are shown in Table 4. Compound 3 strongly inhibited xanthine activity. To the best of our knowledge this is the first report of phenylpropanoid ester glycosides from this plant and hence forms the biochemical and ecological systematic identification. This study therefore validates the folkloric usage of the plant.
CONCLUSION
The study reported the isolation and characterization of secondary metabolites possessing inhibitory activity on xanthine oxidase enzymes implicated in rheumatoid arthritis. The result of the study justified the ethnomedicinal use of the plant in the treatment of inflammatory conditions for which the plant is known and used for.
It is also showed that the compounds isolated and characterized from the plant could be potential anti inflammatory agents.
ACKNOWLEDGMENT
One of the authors (Abiodun Falodun) is grateful to the Third World Academy of Sciences (TWAS) for the Fellowship Award to carry out research at H.E.J. Research Institute of Chemistry, University of Karachi, Pakistan.
REFERENCES
- Ching, F.P., E.K.I. Omogbai, R.I. Ozolua and S.O. Okpo, 2008. Antidiarrhoeal activities of aqueous extract of Stereospermum kunthianum (Chem, Sandrine Petit) stem bark in rodents. Afr. J. Biotechnol., 7: 1220-1225.
Direct Link - Candan, F., 2003. Effect Rhus coriarial L. (Anacardiaceae) on superoxide radical scavenging and xanthine oxidase activity. J. Enzyme Inhibit. Med. Chem., 18: 59-62.
Direct Link - Lildihone, H., D.B. Mauro, H.S. Dulce, C.M. Maria and F. Maysa et al., 2005. Phenylpropanoid glucosides from leaves of Coussarea hydrangeifolia (Rubiaceae). Phytochemistry, 66: 1927-1932.
CrossRef - Lino von Poser, G., J. Schripsena, A.T. Henriques and J.S. Rosendal, 2000. The distribution of iridoids in Bignoniaceae. Biochem. Syst. Ecol., 28: 351-366.
CrossRefDirect Link - Tripetch, K., N. Pawadee, O. Hideaki and R. Somsak, 2006. Lignan, phenolic and iridoid glycosides from Stereospermum cylindricum. Phytochemistry, 67: 516-520.
Direct Link