
Maysa Saleh
Pediatrician, Dubai Health Authority, United Arab Emirates
LiveDNA: 20.32305
Mostafa Helmi
Pediatrician, Dubai Health Authority, United Arab Emirates
Bushra Yacop
Pediatrician, Dubai Health Authority, United Arab Emirates
Pakistan Journal of Biological Sciences
Year: 2020 | Volume: 23 | Issue: 7 | Page No.: 973-976







ABSTRACT
Early infantile epileptic encephalopathy (EIEE) is a severe form neurological disorder of age-related epileptic encephalopathy. Characteristically, it presents with tonic spasms within the first 3 months of life. The spasms can be generalized or focal and hemi-convulsions, it can be in clusters or singly which occur hundreds of times per day, not related to sleep cycle, leading to psychomotor impairment and death. Some cases of EIEE are due to metabolic disorders or brain malformations that may or not be genetic in origin. The genetic origin of EIEE are usually related to brain dysgenesis or neuronal dysfunction. Early infantile epileptic encephalopathy-39 (EIEE39) is a result of homozygous mutation in the SLC25A12 gene (603667) on chromosome 2q31. Here it was described a homozygous nonsense variant of the SLC25A12 gene in our 7 years old child, which was not reported in the literature so far.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Maysa Saleh, Mostafa Helmi and Bushra Yacop, 2020. A Novel Nonsense Gene Variant Responsible for Early Infantile Epileptic Encephalopathy Type 39: Case Report. Pakistan Journal of Biological Sciences, 23: 973-976.
DOI: 10.3923/pjbs.2020.973.976
URL: https://scialert.net/abstract/?doi=pjbs.2020.973.976
DOI: 10.3923/pjbs.2020.973.976
URL: https://scialert.net/abstract/?doi=pjbs.2020.973.976
INTRODUCTION
Early infantile epileptic encephalopathy is a neurological disorder characterized by seizure. Age presentation of EIEE is as early as neonatal period. Usual onset is at the age of 3 months. Neonates are usually presented with hypotonia, generalized and symmetrical tonic spasms, which appear in clusters or singly, may occur hundreds of times per day, independent to sleep cycle1. EIEE encompasses intractable seizures, which may result in devastating global developmental delay and intellectual disability and encephalopathy. This clinical course is usually associated with concomitant brain growth arrest resulting in microcephaly and brain atrophy2.
EIEE has been estimated at 1/100 000 births in Japan and 1/50,000 births in the UK3. EIEE is a genetically heterogeneous epileptic phenotype. Interestingly, genetic etiology is only identified in half of the cases, typically in the form of de novo dominant mutations4.
Early infantile epileptic encephalopathy-39 (EIEE39) is an autosomal recessive, caused by homozygous mutation in the SLC25A12 gene5.
SLC25A12 gene encodes aralar, a neuronal-specific protein localizes to the mitochondria taking part as a malate/aspartate shuttle, AGC1 an aspartate-glutamate carrier isoform, this component is crucial for mitochondrial oxidation of the cytosolic NADH in the brain5.
Case presentation: A 7years old girl known case of cerebral palsy (CP) with epilepsy and developmental delay presented to our ED with cough, difficulty in breathing and increased secretions one day prior to admission especially after feeding. The child developed fever in the ED of 38.4C.
There was history of contact with brother who has URTI symptoms. She is taking her regular Anti-epileptic medications and has been followed up regularly at pediatric Neurology clinic.
She was born to first cousin consanguineous parents. She has severe global developmental delay, no speech development, infantile poorly controlled convulsions (started at age of 40 days), hypertonia, subtle features and persistent lactate elevation. Her deceased brother was similarly affected.
On examination: She looks fatigued, distressed, tachypnea and tachycardia noted.
Chest examinations: Revealed decreased air entry with wheezes, rales and subcostal retraction.
Initial laboratory test reported respiratory acidosis with normal CBC, CRP and electrolytes.
Chest x-ray: Bilateral infiltrates. Managed accordingly with antibiotics.
Over the next few days, the child was clinically deteriorating and required ICU admission with ventilator support.
Cultures revealed MRSA pneumonia, Pseudomonas and fungal infections. Antibiotic adjusted accordingly. Positive RSV nasopharyngeal swab was also noted.
She had persistent acidosis with elevated lactate, despite high dose of sodium bicarbonate IV infusion. Initial metabolic testing including amino-acid chromatography and acylcarnitine as well as urine organic acid reported were normal. The laboratory and clinical findings do not go for the diagnose of a specific genetic disorder, however, for further delineation whole exome sequencing requested.
MRI brain showed diffuse brain atrophy, cortical >white matter, is present with secondary dilation of CSF spaces. The corpus callosum is abnormally thin, otherwise, completely present. The rest of the scan was normal as presented in Fig. 1a, b.
The child condition did not showed any improvement, she had persistent hypotension despite being on inotropes, persistent fever despite antibiotic and anti-fungal coverage, progressive respiratory deterioration despite supportive ventilation. The child developed generalized anasarca with bilateral pleural effusion, low albumin, low platelets and persistent metabolic acidosis. The pleural effusion on the right side was not significant to cause lung collapse. Glasgow Coma Scale Score: 5/15. Repeated investigations revealed pancytopenia (Severe Leukopenia Neutropenia, Thrombocytopenia and Low Hgb). She developed all the picture of secondary hemophagocytic lympho-histiocytosis (HLH)-secondary to infection where there was hypertriglyceridemia, high ferritin levels, low fibrinogen and High LDH, hence managed accordingly.
Whole exome sequencing identified a homozygous non sense variants of c.400>Tp. (Arg134*) (chr2: 172700944, hg 19) in the SLC25A12 gene chromosome 2q31.1. This variant leads to a premature stop codon and subsequent mRNA degradation (nonsense- mediated decay) or truncation of the protein. This was likewise affected her brother who deceased earlier.
Parallel analysis of parental WES data revealed that both parents are heterozygous carriers of the detected variant in the SCL25A12 gene. This confirm homozygosity of the detected variant in our patient as well as her deceased brother.
This variant has previously not been described so far. No allele frequencies in the general population have been documented before.
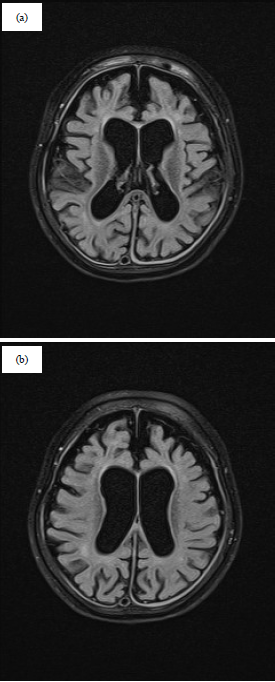 | |
| Fig. 1(a-b): | T2 flair showing diffuse brain atrophy, cortical >white matter, is present with secondary dilation of CSF spaces. The corpus callosum is abnormally thin |
The child was under multidisciplinary management of general pediatrician, nephrologist, hematology, neurology as well as genetic in the PICU. Unfortunately, the child did not survive.
DISCUSSION
Here, a 7 years old girl who has severe global developmental delay and infantile poorly controlled convulsions (started at age of 40 days) was reported. Her whole exome sequencing reveal novel homozygous non-sense variants in SLC25A12 gene chromosome 2q31.1. Her parents were heterozygous carrier to the same variant.
Infantile epileptic encephalopathies (EIEEs) is one of the most severe and earliest form of epilepsy6. It is a heterogeneous group of disorders characterized by intractable seizures and unremitting interictal paroxysmal epileptiform activity, which consequently impair neuro developmental outcomes, during the first year of life1. Onset of EIEE occurs within the first 3 months of life or even within the first few weeks after birth.
Neonates have poor suckling reflexes, hypotonia and manifest with generalized and symmetrical tonic spasms. These spasms are sometimes in cluster or single, lasts up to 10 sec, which may recur hundreds of time a day6. The spasms’ pattern remains unchanged during wakefulness and sleep. Our patient started to have seizure episodes by the age of 40 days.
EIEE may have different etiologies. Many cases have been associated with structural brain abnormalities. Some cases are due to metabolic disorders or brain malformations (such as porencephaly, hemimegalencephaly) that may or not be genetic in origin. Genetic causes should be considered in the absence of structural brain abnormalities or inborn errors of metabolism7. The genetic abnormalities are thought to lead to EIEE as they are related to neuronal dysfunction or brain dysgenesis.
Genetic variants of EIEE have been associated with mutations in certain genes such as ARX (Xp22.13), CDKL5 (Xp22), SL25A22 (11p15.5) and STXBP1 (9q34.1), among others5-8. The wide range of phenotype and genotype heterogeneity makes it difficult to predict with certainty the potentially responsible gene for many EIEEs.
Early infantile epileptic encephalopathy-39 (EIEE39) is caused by homozygous mutation in the SLC25A12 gene (603667) on chromosome8 2q31.1.
Here a homozygous nonsense variant of the SLC25A12 gene was described, which was not reported in the literature so far. This variant leads to a premature stop codon and subsequent mRNA degradation (nonsense- mediated decay) or truncation of the protein. This was likewise affected her brother who deceased.
Parallel analysis of parental WES data revealed that both parents are heterozygous carriers of the detected variant in the SCL25A12 gene. This confirm homozygosity of the detected variant in our patient as well as her deceased brother.
To date, there is no cure for EIEE apart from constant supervision and care. Antiepileptic drugs such as benzodiazepines, valproate, levetiracetam, zonisamide and phenobarbital have shown limited effect in seizures control as has pyridoxine. A ketogenic diet was tried with some success in controlling seizure. Patients with associated metabolic disorders, showed an improvement in the course of EIEE once these conditions have been treated. Similarly, EIEE patients with certain structural abnormalities have benefited from surgical intervention, if unilateral.
CONCLUSION
This report discovers a novel homozygous nonsense variant of the SLC25A12 gene in 7 years old child, which was not reported in the literature so far. That can be beneficial for the establishment of a correct molecular diagnosis for these patients with early infantile epileptic encephalopathy.
SIGNIFICANCE STATEMENT
Though there is no known cure for EIEE apart from supervision and care, yet the establishment of a correct molecular diagnosis has important practical applications as well as significant emotional impact for these patients and parents. In addition, the specific diagnosis can influence treatment decisions later in lifetime. This report will help the researcher to uncover the critical areas of such rare disease that many researchers were not able to explore. Emphasizing, that whole exome sequencing is becoming a powerful tool to accelerate genetic diagnosis as well as for phenotypic expansion of associated clinical manifestations.
REFERENCES
- Asher, Y.J.T. and F. Scaglia, 2012. Molecular bases and clinical spectrum of early infantile epileptic encephalopathies. Eur. J. Med. Genet., 55: 299-306.
CrossRefDirect Link - Sharma, S. and A.N. Prasad, 2013. Genetic testing of epileptic encephalopathies of infancy: An approach. Can. J. Neurol. Sci., 40: 10-16.
CrossRefDirect Link - Kato, M., S. Saitoh, A. Kamei, H. Shiraishi and Y. Ueda et al., 2007. A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet., 81: 361-366.
CrossRefDirect Link - Nashabat, M., X.S. Al Qahtani, S. Almakdob, W. Altwaijri and D.M. Ba-Armah et al., 2019. The landscape of early infantile epileptic encephalopathy in a consanguineous population. Seizure, 69: 154-172.
CrossRefDirect Link - Falk, M.J., D. Li, X. Gai, E. McCormick and E. Place et al., 2014. AGC1 Deficiency Causes Infantile Epilepsy, Abnormal Myelination and Reduced N-Acetylaspartate. In: JIMD Reports, Volume 14, Zschocke, J., K. Gibson, G. Brown, E. Morava and V. Peters (Eds.)., Springer, Berlin, Heidelberg, ISBN: 978-3-662-43747-6, pp: 77-85.
Direct Link - Palmieri, F., 2004. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflügers Archiv., 447: 689-709.
CrossRefDirect Link - Saitsu, H., M. Kato, T. Mizuguchi, K. Hamada and H. Osaka et al., 2008. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat. Genet., 40: 782-788.
CrossRefDirect Link - Mastrangelo, M. and V. Leuzzi, 2012. Genes of early-onset epileptic encephalopathies: From genotype to phenotype. Pediatr. Neurol., 46: 24-31.
CrossRefDirect Link