
Ahmed Gaber
Department of Biology, Faculty of Science, Taif University, P.O. Box 888, Zip code 21974, Taif, Kingdom of Saudi Arabia
LiveDNA: 20.25440
Abdulla A. Alharthi
Department of Pediatrics, Faculty of Medicine, Taif University, Taif, Saudi Arabia
Mohamed M. Hassan
Department of Biology, Faculty of Science, Taif University, P.O. Box 888, Zip code 21974, Taif, Kingdom of Saudi Arabia
Ehab I. El-Hallous
Department of Biology, Faculty of Science, Taif University, P.O. Box 888, Zip code 21974, Taif, Kingdom of Saudi Arabia
Mohamed W. Abukhatwah
Department of Pediatrics, Alhada Armed Forces Hospital, Taif, Saudi Arabia
Abeer M. Almalki
Department of Pediatrics, Children Hospital, Taif, Saudi Arabia
Abdullah A. Muzallef
Abha Maternity Children Hospital, Abha, Saudi Arabia
Saqer Alotaibi
Department of Biotechnology, Faculty of Sciences, Taif University, Taif, Saudi Arabia
LiveDNA: 966.26553
Bandar Aljuaid
Department of Biotechnology, Faculty of Sciences, Taif University, Taif, Saudi Arabia
Walaa F. Alsanie
Department of Clinical Laboratories, Faculty of Applied Medical Sciences, Taif University, Saudi Arabia
Pakistan Journal of Biological Sciences
Year: 2020 | Volume: 23 | Issue: 3 | Page No.: 365-374







ABSTRACT
Background and Objective: Nephrotic syndrome (NS) is the most well-known glomerular kidney disease in children. Genetic risk is highly frequently detected amongst childhood steroid-resistant NS (SRNS) disease. Therefore, the aim of the present research was to evaluate ACTN4 and PLCE1 genes mutations in children with SRNS in western region of Saudi Arabia. Materials and Methods: Twenty SRNS patients were identified and screened for gene mutations within ACTN4 (21 exons) and PLCE1 (33 exons) genes by direct sequencing method to elucidate the correlation between these genes and SRNS. Results: Three novel heterozygous missense mutations were detected in ACTN4 gene in 3 patients. The renal biopsy findings for the 3 patients were focal segmental glomerulosclerosis. The in-silico analysis for these mutations propose that they are deleterious. In case of PLCE1 gene, 2 missense mutations were detected in exons 24 and 26. These mutations were found previously as non-disease-causing mutations. Conclusion: ACTN4 is a significant causative gene mutation of FSGS in Saudi children patients with SRNS But causative mutations in PLCE1 gene can not identified.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Ahmed Gaber, Abdulla A. Alharthi, Mohamed M. Hassan, Ehab I. El-Hallous, Mohamed W. Abukhatwah, Abeer M. Almalki, Abdullah A. Muzallef, Saqer Alotaibi, Bandar Aljuaid and Walaa F. Alsanie, 2020. Screening of ACTN4 and PLCE1 Genes Mutations in Saudi Children Patients with Steroid Resistant Nephrotic Syndrome. Pakistan Journal of Biological Sciences, 23: 365-374.
DOI: 10.3923/pjbs.2020.365.374
URL: https://scialert.net/abstract/?doi=pjbs.2020.365.374
DOI: 10.3923/pjbs.2020.365.374
URL: https://scialert.net/abstract/?doi=pjbs.2020.365.374
INTRODUCTION
Nephrotic syndrome (NS) is the famous glomerular kidney disease in children that is characterize with proteinuria, hypoalbuminemia and edema1. Even though NS is accompanying with several forms of renal disease, the most frequent type (>90%) in pediatric is idiopathic NS. The worldwide incidence of NS in children is reported to be 2-16.9/100,000 children2. Until now, the treatment by corticosteroid remains the most typical treatment for NS patients. Therefore, the types of NS were classified, according to patient's reaction to steroid therapy as steroid-sensitive NS and steroid-resistant NS (SRNS)3. Several reports documented that about 50% of SRNS patients are developing to end-stage renal disease (ESRD) within 15 years4-6. In addition, only around 8-10% of inherited genetic NS is responsive to steroid therapy, which consequently means 90% of SRNS patients are presumed to be multidrug resistant6,7. As stated by the International Study of Kidney Disease in Children, from the pathological point of view, 75% of SRNS patients have focal segmental glomerulosclerosis (FSGS), whereas 20% exhibit minimal change nephrotic syndrome8.
The main reason for the familiar phenotype of SRNS is genetic background, whether recessive or dominant genes9. Consequently, recent studies in SRNS patients discovered single rare mutations in podocyte genes that responsible for SRNS10,11. Sadowski et al.12 reported in 2015 the rate of single-gene causation of SRNS was 29.5% of all tested cases. Recent advances in histology and molecular facilities extended the consideration to the detection of the causative, especially at the molecular level, of SRNS cases. Numerous contributing genes causing NS disease have been distinguished by either utilizing the direct DNA sequencing technique or next-generation sequencing innovation. For example, recessive mutation in NPHS1, NPHS2, LAMB2 or WT1 genes is responsible for about 85% of SRNS patients with onset by three months of age and 66% with onset by 1st year of age13. Additionally, single-gene mutation causing SRNS later in life is linked with the late onset of proteinuria and the disease progression to ESRD14-16. To date, with the recent advances of genome studies, there are over 30 single genes associated with SRNS in children17. Of these, the recognized podocyte genes clarify about 20-30% of hereditary NS and 10-20% of sporadic patients18.
The ACTN4 gene was firstly mapped to chromosome 19q13 and encodes for α-actinin-4 protein, an actin bundling protein of the cytoskeleton that is highly expressed in podocytes19. ACTN4 has a significant role for the cytoskeletal function of the podocyte, with several reports documenting that the knock-down or over expression of the transgenic mouse model with ACTN4 is associated with proteinuria and podocyte alterations20,21. The mutations in ACTN4 were recognized as autosomal dominant late onset causing FSGS19. The PLCE1 gene is mapped to chromosome 10q23-q24 and encodes for the enzyme phospholipase C epsilon-1. The PLCE1 enzyme has a significant role with intracellular signal transduction, therefore, it is extensively expressed in numerous tissues including podocytes22. Several gene mutations in PLCE1 were documented as a reason for autosomal recessive diffuse mesangial sclerosis (DMS)22. In addition, it was found that low protein expression of PLCE1 is correlated with development of podocyte foot process effacement and edematous outer appearance of the zebrafish, elucidating the precise function of the PLCE1 enzyme in the maintenance of the glomerular filtration barrier22. Moreover, PLCE1 gene mutations were considered the major reason for isolated DMS in children, with a rate of 28.6%23. On other hand, Gilbert et al.24 demonstrated that the homozygous mutation in PLCE1 is not always enough to cause DMS. The percentage of consanguineous marriage in the Kingdom of Saudi Arabia (KSA) was reported to be around 56%25,26. Pediatric renal diseases are more likely to be predominant in countries with high percentages of consanguineous marriages, such as KSA27. Furthermore, congenital and infantile NS are documented with a higher incidence in KSA than in other countries28.
Previously, same Saudi children patients with SRNS that are presented here were screened the for mutation analysis within the NPHS2 gene and exons 8 and 9 of the WT1 gene29. The gene mutations of NPHS2 gene are present in 10% of the patients, while WT1 gene mutation is found in 5%29. To further evaluate the existence of single gene mutation within the same cohort, mutational screening of all exons of ACTN4 and PLCE1 genes was achieved. Until now, this study is consider the first detection of ACTN4 gene mutation in Saudi children with SRNS.
MATERIALS AND METHODS
Study area: This study was carried out at molecular genetics Lab, Deanship of Scientific Research, Taif University, KSA from January, 2018-April, 2019. The study was approved by the bioethics committee of Al-Hada Armed Forces Hospital (PTRC#15-05-227). Twenty children who entered Al-hada Armed Forces Hospital during the period of 2015-2017 with age between 1 and 16 years old and with a clinical finding of SRNS were enrolled in this study, participation involved informed consent. NS cases were characterized as proteinuria, hypoalbuminemia and generalized edema30. SRNS patients were described as the inability to respond to daily treatment of prednisone dose (2 mg kg–1) for a period of 4-6 weeks30. SRNS patients were described as the inability to response to daily treatment of prednisone dose (2 mg kg–1) for a period of 4-6 weeks30. The inclusion criterion comprised all Saudi childhood SRNS patients. Exclusion criteria were steroid sensitive NS patients and/or patients with secondary reason (e.g., IgA nephropathy). The renal biopsy samples were achieved by renal pathologists. Several clinical analysis were recorded, such as: sex, family history, age of onset, hypertension, hematuria, failure to the response to steroid therapy, kidney biopsy and interval time of development to ESRD.
Genomic DNA extraction: Total DNA was extracted immediately from blood by Genomic DNA Purification Kit (Promega, USA) as described by the protocol of the manufacture. The purity and concentration of the genomic DNA was evaluated through agarose gel electrophoresis. All coding sequence and exon-intron borders of both genes, ACTN4 (accession number NM_004924.5, 21 exons) and PLCE1 (accession number NM_016341.3, 33 exons), were amplified by PCR using specific primers as previously reported by Dai et al.31 and Boyer et al.32. PCR reaction mixture and conditions were performed as reported previously by Dai et al.31 and Boyer et al.32.
Detection mutations by PCR-restriction fragment length polymorphism (RFLP) analysis: The two mutations that were found in the PLCE1 gene (c.5964C>T in exon 24, c.6414A>G in exon 26) were confirmed using the PCR/RFLP technique. PCR assay was conducted using specific primers for exon 24 and exon 26 as described previously by Dai et al.31 and Boyer et al.32. PCR reaction mixture of the final volume of 50 μL included: 50 ng of genomic DNA, 0.2 μM of forward and reverse primer and 25 μL of GoTaq® green master mix, with the remaining volume comprising Nuclease-Free Water (Promega, USA). The RFLP assay was carried out with the BseYI enzyme (New England Biolabs, USA) in order to detect c.5964C>T mutation in exon 24 (544 bp), while the BstUI enzyme (New England Biolabs, USA) was used to detect c.6414A>G mutation in exon 26 (488 bp). Conditions of digestion with restriction enzymes were achieved as described by the manufacture’s protocol.
In-silico analysis of the mutated nucleotides: Expectation of the functional influence of each pathogenic mutation found in ACTN4 and PLCE1 genes was achieved using 3 distinctive computational programs freely accessible online, (1) The PROVEAN program (http://provean.jcvi.org/seq_ submit.php) analyzes the single nucleotide polymorphisms and/or indels33. As described by PROVEAN, a value of -2.5 is assumed to be a default threshold. In this way, mutations with a score equivalent to or lower than -2.5 are considered deleterious. On other hand, mutations with a score higher than -2.5 are considered neutral, (2) The PolyPhen2 program (http://genetics.bwh.harvard.edu/pph2/) employs the sequence homology and information of the 3D structure of the protein to anticipate the likely influence of an amino acid substitution on the structure and function of a human protein. The results are categorized as benign, possibly damaging, probably damaging or unknown34, (3) The SIFT program, (https://sift.bii.a-star.edu.sg/www/SIFT_seq_submit2.html) assesses whether the replacement of an amino acid disturbs protein function35. To further explore the impact of all mutations found in this study on the 3D structure and folding of ACTN4 and PLCE1 proteins, the active domains of both proteins were modeled using Protein Fold Recognition Server (Phyre2)36 and the structure of domains and mutation site were visualized using PyMol37.
Statistical analysis: Sequencing of the purified specific fragments were performed as previously described29. The raw sequencing data were assembled by the Data collection software version 3.1 (Applied Biosystems). Sequence base-calling and point mutation analysis were investigated using Seqscape software version 2.7 (Applied Biosystems). Mutational records were named through the nomenclature of the Human Genome Variation Society38.
RESULTS
Effect of ACTN4 gene mutation on protein structure: The summary of clinical data of the individual cases that have the mutations of ACTN4 and PLCE1 genes is shown in Table 1. The reference sequences of the ACTN4 gene are NG_007082.2, NM_004924.5 and NP_004915.2. Three novel mutations were detected in ACTN4 gene in three SRNS patients as heterozygous state (Fig. 1). The first one found in exon 8 (c.870T>A, p.V247E). The second mutation (c.2136T>A, p.M669K) was in exon 16, while the third mutation (c.2464C>A, p.D778E) was in exon 18. The three mutations occurred in 3 distinct domains of the ACTN4 gene (Fig. 1a, b). On the level of the 3D protein structure and function, the three mutations were found to affect the structure and polar charge of the amino acid residues that have a significance for the coiled coil structure (Fig. 1c).
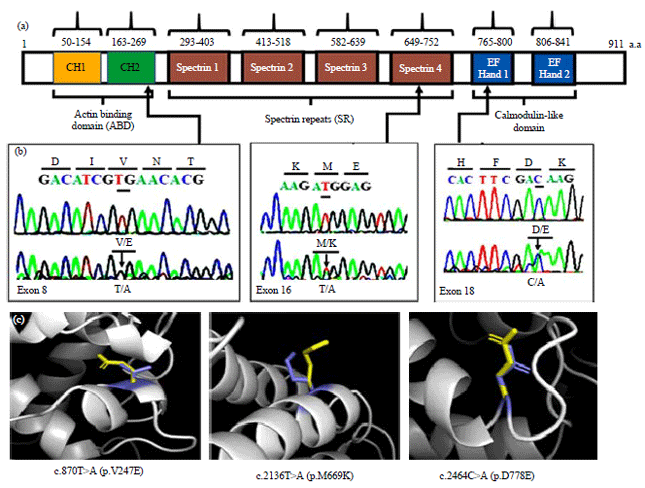 | |
| Fig. 1(a-c): | DNA sequence analysis of ACTN4 gene, (a) Functional different domains of the human ACTN4 protein, (b) Direct sequencing of the PCR product amplified from exons 8, 16 and 18 of ACTN4 gene. The affected individuals are heterozygous for the 3 mutations (arrow) and (c) Superimposed tertiary structure of ACTN4 protein and the predictions of normal and mutated amino acid |
Normal amino acid is showed in purple, while the mutated amino acid in the three mutations is illustrated in yellow | |
| Table 1: | Clinical data of the individual children patients with ACTN4 and PLCE1 gene mutations |
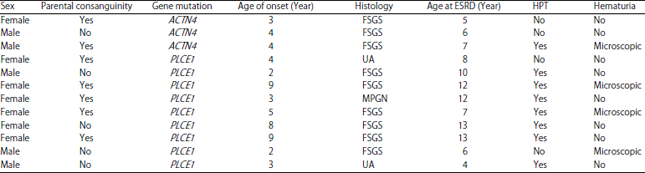 | |
FSGS: Focal segmental glomerulosclerosis, HPT: Hypertension, MPGN: Membranoproliferative glomerulonephritis, UA: Unavailable, ESRD: End stage renal disease | |
The coiled coil structure has a significant function in key cellular interactions. The replacement of the amino acids valine and methionine by glutamic acid and lysine in p.V247E and p.M669K mutations, respectively, altered the surface charge of the amino acids in this area from non-polar to negative and positive charges, respectively. Consequently, the three mutations of ACTN4 gene could be deleterious within the SRNS patients.
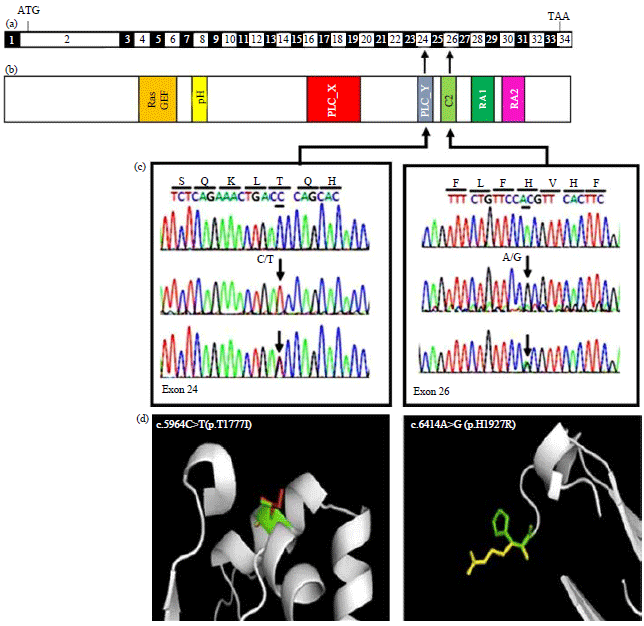 | |
| Fig. 2(a-d): | DNA sequence analysis of PLCE1 gene, (a) Exon structure of human PLCE1 gene-viewing positions of start and stop codons, (b) Locations of the protein domains regarding the encoding exon site, (c) Direct sequencing of the PCR product amplified from exons 24 and 26 PLCE1 gene. Arrows indicate relative positions of mutations and (d) Superimposed tertiary structure of PLCE1 protein and the predictions of normal and mutated amino acid |
Normal amino acid is showed in green, while the mutated amino acid in c.5964C>T mutation is illustrated in red and in c.6414A>G is showed in yellow | |
Mutation screening of PLCE1 gene: The reference symbols of the PLCE1 gene and protein are NG_015799.1, NM_016341.3 and NP_057425.3. Two missense mutations were detected in PLCE1 gene in 2 different domains (Fig. 2a, b). First one at the position c.5964C>T (p.T1777I) in exon 24 and the other at the position c.6414A>G (p.H1927R) in exon 26 (Fig. 2c). These two variants were found as homozygous, heterozygous and compound mutation states in 9 out of 20 (45%) SRNS patients. Unfortunately, parent’s DNA of the compound mutations were not analyzed to investigate if these variants are located on one chromosome and whether, therefore, the compound mutations would be characterized as a haplotype heterozygous mutation or if they are located on two separate chromosomes and they would be compound heterozygous mutations. The amino acid Isoleucine and Arginine that replace Tyrosine and Histidine in the two mutations have a different polar structure (Fig. 2d).
PCR-RFLP analysis of PLCE1 gene mutations: By using BseYI restricted enzyme, The c.5964T>C mutation in exon 24 display one band at 544 base-pair in homozygous carrier patient, while 3 bands were observed in the patients with heterozygous state for the mutation (Fig. 3a).
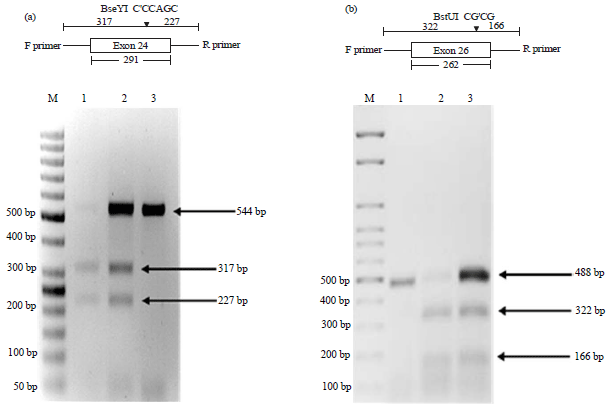 | |
| Fig. 3(a-b): | PCR/RFLP analysis confirmation of PLCE1 gene mutations, (a) Schematic diagram and gel electrophoresis of c.5964T>C mutation after cutting exon 24 with BseYI enzyme and (b) Schematic diagram and gel electrophoresis of c.6414A>G mutation after cutting exon 26 with BstUI enzyme |
a: M: 50 bp marker, 1: Wild type exon, 2: Heterozygous mutated exon, 3: Homozygous mutated exon, b: M: 100 bp marker, 1: Wild type exon, 2: Heterozygous mutated exon, 3: Homozygous mutated exon | |
| Table 2: | In-silico analysis of the missense mutations identified in ACTN4 and PLCE1 genes |
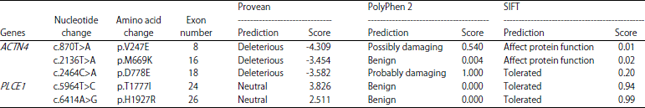 | |
In case of c.6414A>G mutation in exon 26, the BstUI restriction enzyme was used in order to detect the carrier patients with mutations. Thus, the samples from patients with homozygous state generated 2 bands, whereas, three bands were detected in all patients that have the heterozygous state (Fig. 3b).
In-silico evaluation of ACTN4 and PLCE1 genes mutations: The mutation of p.V247E in exon 8 was predicted to be possibly damaging and consequently affect the function of the ACTN4 protein (Table 2). Thus, the amino acid valine at the position 247 may be functionally significant and the mutation may lead to damaging the interference with conformation and function of the ACTN4 protein. Additionally, the 2 mutations p.M669K and p.D778E in ACTN4 gene were found to be possibly deleterious by PROVEAN software (Table 2). The variation p.D778E was predicted to be probably damaging and tolerated by the PolyPhen2 and SIFT programs, respectively. In case of PLCE1 gene, the two mutations were predicted to be benign by the three web-based programs (Table 2).
DISCUSSION
NS is one of the most widely recognized reasons for kidney disease in children1. To date, numerous genes have been associated with NS in children, which is indicative of the wide genetic cause of this syndrome17. Mutation screening in SRNA patients would be beneficial for clinicians to minimize the risk of immunosuppressive drugs, since gene mutations are involved with a poor response to immunosuppression39,40.
The current study presents the first molecular screening analysis for mutations of the ACTN4 gene in a cohort of Saudi child patients with SRNS. The mean age of the present patients with SRNS at the onset of diagnosis was 4.5 years. Similar results were published recently of childhood NS in tropical Africa with a mean age of 4.8 years41 and 4.0 years in Brazilian children42. On other hand, the mean age at the onset of diagnosis was 5.4 years in Indian children43 and 2.3 years in African American child patients44.
There is a paucity in the molecular research for screening mutations of genes that are related to SRNS, especially in Saudi society. In a recent study, mutation screening for the complete 8 exons of the NPHS2 gene and exons 8 and 9 of the WT1 gene were performed using the same number of child patients that are represented here29. Additionally, Al-Hamed et al.45 detected mutations in NPHS1, NPHS2, PLCE1 and MYO1E genes in a group of 49 Saudi families having congenital NS, infantile NS and childhood SRNS. Furthermore, Kari et al.46 reported a lower frequency rate for the mutations in NPHS2 and NPHS1 genes in Saudi child patients with SRNS. More recently, Hashmi et al.47 detected the cause of SRNS in a consanguineous Saudi family by detecting a homozygous novel insertion mutation (c.6272_6273insT) in the PLCE1 gene using whole exome sequencing. Therefore, this study presented the first mutation screen analysis of the ACTN4 gene for Saudi children with SRNS. Molecular screening analysis revealed three mutations in the ACTN4 gene that were found in three patients with a frequency of 15% (3/20) in the heterozygous state. These mutations are not recognized in the db SNP, 1000G and ExAC databases and are anticipated to be deleterious and disease-causing by PROVEAN, Polyphen2 and SIFT programs (Table 2). The first mutation p.V247E is located in the actin-binding domain (Fig. 1). To date, all mutations that were identified in the ACTN4 gene have been found in the actin-binding domain of the encoded protein, resulting in irregular actin rich cellular aggregates48. The recognizable proof of ACTN4 mutations as a reason for human kidney disease proves a significant cellular pathway of the ACTN4 mutations and podocyte dysfunction49.
The renal biopsy pattern for the three patients having ACTN4 gene mutations is FSGS that progressed to ESRD after 2-3 years from the onset of the diagnosis (Table 1). These results agree with a clinical study reported by Choi et al.50, in which they found the mutation of ACTN4 gene at position S262F as a heterozygous form was connected with FSGS at age 3 to 4 years old and directly progressed to ESRD. Additionally, three different mutations occurring in ACTN4 in three different families were the cause of FSGS20. The incidence of FSGS has increased remarkably in the past 20 years and accounted for 50-60% of the total kidney biopsies in childhood SRNS51. There is a close relationship between the single gene mutations of the podocyte proteins, such as NPHS1, NPHS2, ACTN4, LAMB2, INF2, CD2AP or TRPC6 and the FSGS pattern52. By immunofluorescence staining, it was documented that the ACTN4 protein is dispersed in podocytes, with a little spreading in another vasculature in the renal cortex. These results confirmed the reports of the high expression of ACTN4 protein in the kidney53.
It is difficult to estimate whether the novel mutations of ACTN4 gene are the reason for the SRNS in the three patients without examining the family pedigree and segregation. However, in-silico programs have been used to identify the pathogenicity of these novel mutations (Table 2). Several reports used the same in-silico programs to anticipate the effect of gene mutations within SRNS patients45,54,55. Furthermore, the effect of the three mutations on the 3D structure of the ACTN4 protein indicated that the three mutations disturb the coiled coil alpha helix in three domains of the ACTN4 protein (Fig. 1c). These three domains are important for the function of ACTN4. In addition, the sequence alignment of the ACTN4 protein with other proteins from different organisms showed that about 99% of all amino acids in theses domains are conserved (data not shown). Consequently, a mutation in these domains allowed to change the protein structure has to affect the original function of this protein.
According to the literature, the 2 mutations (p.T1777I and p.H1927R) found in the PLCE1 gene were non-pathogenic variants and considered as single nucleotide polymorphisms39. Interestingly, in several reports, same variants were found to associate with the risk of cancer. Malik et al.56 documented that these mutations were significantly linked to the elevated risk of esophageal cancer in Kashmir Valley. Moreover, Wang et al.57 confirmed that the variant p.H1927R might play an essential role in esophageal carcinogenesis in the Chinese population by changing the protein structure and enzyme activity and thus expanding the inflammatory process in esophageal epithelium. Consequently, Wang et al.57 conclude that the p.H1927R variant may establish a promising biomarker for esophageal cell carcinoma risk stratification, early detection and progression prediction. Moreover, Yuan et al.58 found that p.H1927R was remarkably linked with the increased risk of head and neck cancer in Chinese populations. All these results suggest the association of the 2 variants of the PLCE1 gene with different types of cancer. Therefore, the 2 variants of the PLCE1 gene might be used as a biomarker for childhood SRNS.
The present study has some limitations. The number of patients is small, the inability to identify the compound mutations and the inability to include a control group. However, a relationship between single gene mutation in ACTN4 gene and Saudi child patients with SRNS might be established.
CONCLUSION
The present study investigates causative mutations linked with SRNS by screening 2 well-known genes (ACTN4 and PLCE1). Among 20 children patients who enrolled for PCR sequencing for all exons of ACTN4 and PLCE1 genes, 3 patients carried three novel heterozygous mutations causing disease in ACTN4 gene. Furthermore, two known variants were detected in PLCE1 gene in nine patients. As indicated by kidney biopsies, there is a high frequency of FSGS among children patients. Identification of mutations causing SRNS is of importance, for helping in therapeutic considerations as well as for genetic advising.
ACKNOWLEDGMENT
The authors appreciatively thank every one of the patients who were tried out this examination. Special thanks to the specialized staff, the nursing and medical staff at the participating hospital for their assistance with enrolment of the patients for the investigation. This study was funded by Deanship of Scientific Research, Taif University, Saudi Arabia (research project number 1-437-5387).
SIGNIFICANCE STATEMENT
The present study elucidated for the first time the existence of the ACTN4 gene mutations in Saudi children patients with steroid resistant nephrotic syndrome. These results are helpful to understand the role of such gene in the development and pathogenesis of SRNS among Saudi children.
REFERENCES
- Rheault, M.N., C.C. Wei, D.S. Hains, W. Wang, B.A. Kerlin and W.E. Smoyer, 2014. Increasing frequency of acute kidney injury amongst children hospitalized with nephrotic syndrome. Pediatr. Nephrol., 29: 139-147.
CrossRefDirect Link - Shatat, I.F., L.J. Becton and R.P. Woroniecki, 2019. Hypertension in childhood nephrotic syndrome. Front. Pediatr., Vol. 7.
CrossRefDirect Link - Gao, X., Y. Ma, L. Sun, D. Chen, C. Mei and C. Xu, 2014. Cyclosporine A for the treatment of refractory nephrotic syndrome with renal dysfunction. Exp. Therapeut. Med., 7: 447-450.
CrossRefDirect Link - Zagury, A., A.L. de Oliveira, J.A.A. Montalvao, R.H.L. Novaes, V.M. de Sa, C.A.P. de Moraes and M. de Sousa Tavares, 2013. Steroid-resistant idiopathic nephrotic syndrome in children: Long-term follow-up and risk factors for end-stage renal disease. Braz. J. Nephrol., 35: 191-199.
CrossRefDirect Link - Trautmann, A., M. Bodria, F. Ozaltin, A. Gheisari and A. Melk et al., 2015. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: The PodoNet registry cohort. Clin. J. Am. Soc. Nephrol., 10: 592-600.
CrossRefDirect Link - Mekahli, D., A. Liutkus, B. Ranchin, A. Yu and L. Bessenay et al., 2009. Long-term outcome of idiopathic steroid-resistant nephrotic syndrome: A multicenter study. Pediatr. Nephrol., 24: 1525-1532.
CrossRefDirect Link - Buscher, A.K., B. Kranz, R. Buscher, F. Hildebrandt and B. Dworniczak et al., 2010. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol., 5: 2075-2084.
CrossRefDirect Link - D'Agati, V.D., 2008. The spectrum of focal segmental glomerulosclerosis: New insights. Curr. Opin. Nephrol. Hypertens., 17: 271-281.
CrossRefDirect Link - Rachmadi, D., A. Melani and L. Monnens, 2015. NPHS2 gene mutation and polymorphisms in Indonesian children with steroid-resistant nephrotic syndrome. Open J. Pediatr., 5: 27-33.
CrossRefDirect Link - Joshi, S., R. Andersen, B. Jespersen and S. Rittig, 2013. Genetics of steroid-resistant nephrotic syndrome: A review of mutation spectrum and suggested approach for genetic testing. Acta Paediatr., 102: 844-856.
CrossRefDirect Link - Sampson, M.G., J.B. Hodgin and M. Kretzler, 2015. Defining nephrotic syndrome from an integrative genomics perspective. Pediatr. Nephrol., 30: 51-63.
CrossRefDirect Link - Sadowski, C.E., S. Lovric, S. Ashraf, W.L. Pabst and H.Y. Gee et al., 2015. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol., 26: 1279-1289.
CrossRefDirect Link - Hinkes, B.G., B. Mucha, C.N. Vlangos, R. Gbadegesin and J. Liu et al., 2007. Nephrotic syndrome in the first year of life: Two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1 and LAMB2). Pediatrics, 119: e907-e919.
CrossRefPubMedDirect Link - McCarthy, H.J., A. Bierzynska, M. Wherlock, M. Ognjanovic and L. Kerecuk et al., 2013. Simultaneous sequencing of 24 genes associated with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol., 8: 637-648.
CrossRefDirect Link - Lovric, S., S. Ashraf, W. Tan and F. Hildebrandt, 2016. Genetic testing in steroid-resistant nephrotic syndrome: When and how? Nephrol. Dial. Transplant., 31: 1802-1813.
CrossRefDirect Link - Sanna-Cherchi, S., K.E. Burgess, S.N. Nees, G. Caridi and P.L. Weng et al., 2011. Exome sequencing identified MYO1E and NEIL1 as candidate genes for human autosomal recessive steroid-resistant nephrotic syndrome. Kidney Int., 80: 389-396.
CrossRefDirect Link - Noone, D.G., K. Iijima and R. Parekh, 2018. Idiopathic nephrotic syndrome in children. Lancet, 392: 61-74.
CrossRefDirect Link - Bierzynska, A., K. Soderquest and A. Koziell, 2015. Genes and podocytes-new insights into mechanisms of podocytopathy. Front. Endocrinol., Vol. 5.
CrossRefDirect Link - Mathis, B.J., S.H. Kim, K. Calabrese, M. Haas, J.G. Seidman, C.E. Seidman and M.R. Pollak, 1998. A locus for inherited focal segmental glomerulosclerosis maps to chromosome 19q13. Kidney Int., 53: 282-286.
CrossRefDirect Link - Kaplan, J.M., S.H. Kim, K.N. North, H. Rennke and L.A. Correia et al., 2000. Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet., 24: 251-256.
CrossRefPubMedDirect Link - Kos, C.H., T.C. Le, S. Sinha, J.M. Henderson and S.H. Kim et al., 2003. Mice deficient in α-actinin-4 have severe glomerular disease. J. Clin. Invest., 111: 1683-1690.
CrossRefDirect Link - Hinkes, B., R.C. Wiggins, R. Gbadegesin, C.N. Vlangos and D. Seelow et al., 2006. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat. Genet., 38: 1397-1405.
CrossRefDirect Link - Gbadegesin, R., B.G. Hinkes, B.E. Hoskins, C.N. Vlangos and S.F. Heeringa et al., 2008. Mutations in PLCE1 are a major cause of Isolated Diffuse Mesangial Sclerosis (IDMS). Nephrol. Dial. Transplant., 23: 1291-1297.
CrossRefDirect Link - Gilbert, R.D., C.L.S. Turner, J. Gibson, P.S. Bass and M.R. Haq et al., 2009. Mutations in phospholipase C epsilon 1 are not sufficient to cause diffuse mesangial sclerosis. Kidney Int., 75: 415-419.
PubMedDirect Link - Al-Abdulkareem, A.A. and S.G. Ballal, 1998. Consanguineous marriage in an urban area of Saudi Arabia: Rates and adverse health effects on the offspring. J. Community Health, 23: 75-83.
CrossRefDirect Link - El Mouzan, M.I., A.A. Al Salloum, A.S. Al Herbish, M.M. Qurachi and A.A. Al Omar, 2008. Consanguinity and major genetic disorders in Saudi children: A community-based cross-sectional study. Ann. Saudi Med., 28: 169-173.
Direct Link - Kari, J.A., D. Bockenhauer, H. Stanescu, M. Gari, R. Kleta and A.K. Singh, 2014. Consanguinity in Saudi Arabia: A unique opportunity for pediatric kidney research. Am. J. Kidney Dis., 63: 304-310.
CrossRefDirect Link - Abdurrahman, M.B., F.H. Shipkey, A.T.H. Elidrissy and W. Al-Kahtani, 1989. Nephrotic syndrome in Saudi infants in the first year of life. Ann. Trop. Paediatr., 9: 140-146.
CrossRefDirect Link - Alharthi, A.A., A. Gaber, M.W. AbuKhatwah, A.M. Almalki and A.A. Muzallef et al., 2017. Mutational analysis of NPHS2 and WT1 genes in Saudi children with nephrotic syndrome. Curr. Pediatr. Res., 21: 11-18.
Direct Link - Dai, S., Z. Wang, X. Pan, X. Chen and W. Wang et al., 2009. ACTN4 gene mutations and single nucleotide polymorphisms in idiopathic focal segmental glomerulosclerosis. Nephron Clin. Pract., 111: c87-c94.
PubMedDirect Link - Boyer, O., G. Benoit, O. Gribouval, F. Nevo and A. Pawtowski et al., 2010. Mutational analysis of the PLCE1 gene in steroid resistant nephrotic syndrome. J. Med. Genet., 47: 445-452.
CrossRefDirect Link - Choi, Y., G.E. Sims, S. Murphy, J.R. Miller and A.P. Chan, 2012. Predicting the functional effect of amino acid substitutions and indels. PLoS One, Vol. 7.
CrossRefDirect Link - Adzhubei, I.A., S. Schmidt, L. Peshkin, V.E. Ramensky and A. Gerasimova et al., 2010. A method and server for predicting damaging missense mutations. Nat. Methods, 7: 248-249.
CrossRefDirect Link - Ng, P.C. and S. Henikoff, 2003. SIFT: Predicting amino acid changes that affect protein function. Nucl. Acids Res., 31: 3812-3814.
CrossRefDirect Link - Kelley, L.A., S. Mezulis, C.M. Yates, M.N. Wass and M.J. Sternberg, 2015. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protocols, 10: 845-858.
CrossRefDirect Link - Preston, R., H.M. Stuart and R. Lennon, 2019. Genetic testing in steroid-resistant nephrotic syndrome: Why, who, when and how? Pediatr. Nephrol., 34: 195-210.
CrossRefDirect Link - McCaffrey, J., R. Lennon and N.J. Webb, 2016. The non-immunosuppressive management of childhood nephrotic syndrome. Pediatr. Nephrol., 31: 1383-1402.
CrossRefDirect Link - Olowu, W.A., A. Ademola, A.B. Ajite and Y.M. Saad, 2017. Childhood nephrotic syndrome in tropical Africa: Then and now. Paediatr. Int. Child Health, 37: 259-268.
CrossRefDirect Link - Guaragna, M.S., A.C.G.B. Lutaif, C.S.C. Piveta, M.L. Souza and S.R. de Souza et al., 2015. NPHS2 mutations account for only 15% of nephrotic syndrome cases. BMC Med. Genet., Vol. 16.
CrossRefDirect Link - Dhandapani, M.C., V. Venkatesan, N.B. Rengaswamy, K. Gowrishankar, S. Ekambaram, P. Sengutavan and V. Perumal, 2017. Report of novel genetic variation in NPHS2 gene associated with idiopathic nephrotic syndrome in South Indian children. Clin. Exp. Nephrol., 21: 127-133.
CrossRefDirect Link - Chernin, G., S.F. Heeringa, R. Gbadegesin, J. Liu and B.G. Hinkes et al., 2008. Low prevalence of NPHS2 mutations in African American children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol., 23: 1455-1460.
CrossRefDirect Link - Al-Hamed, M.H., E. Al-Sabban, H. Al-Mojalli, N. Al-Harbi and E. Faqeih et al., 2013. A molecular genetic analysis of childhood nephrotic syndrome in a cohort of Saudi Arabian families. J. Hum. Genet., 58: 480-489.
CrossRefPubMedDirect Link - Kari, J.A., S.M. El-Desoky, M. Gari, K. Malik, V. Vega-Warner, S. Lovric and D. Bockenhauer, 2013. Steroid-resistant nephrotic syndrome: Impact of genetic testing. Ann. Saudi Med., 33: 533-538.
CrossRefDirect Link - Hashmi, J.A., R.A. Safar, S. Afzal, A.M. Albalawi, F. Abdu-Samad, Z. Iqbal and S. Basit, 2018. Whole exome sequencing identification of a novel insertion mutation in the phospholipase C ε-1 gene in a family with steroid resistant inherited nephrotic syndrome. Mol. Med. Rep., 18: 5095-5100.
CrossRefDirect Link - Feng, D., C. DuMontier and M.R. Pollak, 2015. The role of alpha-actinin-4 in human kidney disease. Cell Biosci., Vol. 5.
CrossRefDirect Link - Feng, D., J. Notbohm, A. Benjamin, S. He and M. Wang et al., 2018. Disease-causing mutation in α-actinin-4 promotes podocyte detachment through maladaptation to periodic stretch. Proc. Natl. Acad. Sci. USA., 115: 1517-1522.
CrossRefDirect Link - Choi, H.J., B.H. Lee, H.Y. Cho, K.C. Moon and I.S. Ha et al., 2008. Familial focal segmental glomerulosclerosis associated with an ACTN4 mutation and paternal germline mosaicism. Am. J. Kidney Dis., 51: 834-838.
CrossRefDirect Link - Bouts, A., F. Veltkamp, B. Tönshoff, M. Vivarelli and Working Group “Transplantation”, “Idiopathic Nephrotic Syndrome” of the European Society of Pediatric Nephrology, 2019. European Society of Pediatric Nephrology survey on current practice regarding recurrent focal segmental glomerulosclerosis after pediatric kidney transplantation. Pediatr. Transplant., Vol. 23, No. 3.
CrossRefDirect Link - Chen, Y.M. and H. Liapis, 2015. Focal segmental glomerulosclerosis: Molecular genetics and targeted therapies. BMC Nephrol., Vol. 16.
CrossRefDirect Link - Gbadegesin, R., B. Bartkowiak, P.J. Lavin, N. Mukerji and G. Wu et al., 2009. Exclusion of homozygous PLCE1 (NPHS3) mutations in 69 families with idiopathic and hereditary FSGS. Pediatr. Nephrol., 24: 281-285.
CrossRefDirect Link - Leong, I.U.S., A. Stuckey, D. Lai, J.R. Skinner and D.R. Love, 2015. Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination and two metaservers to classify long QT syndrome gene mutations. BMC Med. Genet., Vol. 16.
CrossRefDirect Link - Rodrigues, C., A. Santos-Silva, E. Costa and E. Bronze-da-Rocha, 2015. Performance of in silico tools for the evaluation of UGT1A1 missense variants. Hum. Mutat., 36: 1215-1225.
CrossRefDirect Link - Malik, M.A., M. Umar, U. Gupta, S.A. Zargar and B. Mittal, 2014. Phospholipase C epsilon 1 (PLCE1 rs2274223A>G, rs3765524C>T and rs7922612C>T) polymorphisms and esophageal cancer risk in the Kashmir Valley. Asian Pac. J. Cancer Prev., 15: 4319-4323.
CrossRefPubMedDirect Link - Wang, L.D., X. Bi, X. Song, N.M. Pohl and Y. Cheng et al., 2013. A sequence variant in the phospholipase C epsilon C2 domain is associated with esophageal carcinoma and esophagitis. Mol. Carcinog., 52: 80-86.
CrossRefDirect Link - Yuan, Z., H. Yuan, H. Ma, M. Chu and Y. Wang et al., 2013. Genetic variants at 10q23 are associated with risk of head and neck cancer in a Chinese population. Oral Oncol., 49: 332-335.
CrossRefDirect Link