
Zenshiny Starlin
Faculty of Pharmacy, Universitas Indonesia, Depok, West Java, Indonesia
Yahdiana Harahap
Faculty of Pharmacy, Universitas Indonesia, Depok, West Java, Indonesia
LiveDNA: 62.20262
Eme S. Sitepu
Faculty of Pharmacy, Universitas Indonesia, Depok, West Java, Indonesia
Pakistan Journal of Biological Sciences
Year: 2020 | Volume: 23 | Issue: 10 | Page No.: 1321-1331







ABSTRACT
Background and Objective: Acrylamide (AA) is a carcinogenic substance that is easily found in working environment, food, contaminated air and tobacco smoke. This substance can be distributed rapidly through all body compartments. The aim of this study is to get the method for determining acrylamide in dried blood spot. Materials and Methods: Dried blood spot was used as the bio-sampling method and was optimized and validated by using propranolol as the internal standard. The sample was prepared using a protein precipitation technique optimized. Reversed-phase chromatography with Acquity® UPLC BEH C18 column (1.7, 2.1× 100 mm) was used for compound separation. Results: Optimized analytical condition for this substance was eluted with the flow rate of 0.20 mL/min under a gradient of the mobile phase of 0.1% formic acid in water and acetonitrile within 3 min. Triple quadrupole mass spectrometry with electrospray ionization (ESI) in positive mode was used as quantification analysis. The Multiple Reaction Monitoring (MRM) was set at m/z 71.99>55.23 (m/z) for acrylamide and 260.2>116.2 (m/z) for propranolol. The range of concentration was linear within 2.5-100 μg mL–1. Conclusion: All the validation parameters were fulfilled the criteria in US FDA Guideline for Bioanalytical Method Validation 2018.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Zenshiny Starlin, Yahdiana Harahap and Eme S. Sitepu, 2020. Method Validation of Acrylamide in Dried Blood Spot by Liquid Chromatography-tandem Mass Spectrometry. Pakistan Journal of Biological Sciences, 23: 1321-1331.
DOI: 10.3923/pjbs.2020.1321.1331
URL: https://scialert.net/abstract/?doi=pjbs.2020.1321.1331
DOI: 10.3923/pjbs.2020.1321.1331
URL: https://scialert.net/abstract/?doi=pjbs.2020.1321.1331
INTRODUCTION
Acrylamide (AA) is a carcinogenic compound. This compound is found in the working area, food, drinking water, contaminated air and tobacco smoke1,2. A high level of acrylamide is in food that contains a high level of carbohydrate and protein, which is going through fried, baked and another heating process. Crisp fried potato, popcorn, biscuit, bread, chocolate powder, coffee powder and beer contain high-level acrylamide2,3. The comparison of acrylamide levels between smoking and non-smoking laboratory workers shows a significant difference in result. The workers who smoke have 54 pmoL g–1 acrylamide. On the other hand, the workers who do not smoke have 31 pmoL g–1 acrylamide4. Each tobacco contains 2.40 μg acrylamide5.
Acrylamide is rapidly absorbed into systemic and then distributed to all tissue. These substances are going through metabolism in the body and excreted from urine. This compound interacts with protein and DNA forming a DNA adducts6. Some acrylamides is going through phase one metabolism converted to glycidamide7–9. This compound also interacts with nitrogen of DNA base forming DNA adducts. Acrylamide has a reactivity mechanism with three different pathways. The pathways include the polymerization of acrylamide to polyacrylamide, the carbanion addition to positions α and β form genotoxic acrylamide10,11 and the acrylamide oxidation into epoxide metabolite, glycidamide9,10.
The determination of AA levels in biological fluids is essential because of the potential risk of AA exposure. Methods for AA quantification in biological fluids have been carried out in different biological matrices12–14. The toxicokinetic tests have also been carried out in human and rat serum15. However, the research uses venipuncture as a method of bio-sampling and d3-acrylamide as an internal standard12,15. The venipuncture method has disadvantages such as; a problematic protocol for obtaining, storing and distributing blood. This method also requires a certified phlebotomist, lots of equipment and gives patients pain16–18. The labeled radioisotope used as an internal standard is very expensive. Isomer or another compound that has the same characteristic with AA can replace the radioisotope's internal standard. The methods test parameters refers to US FDA’s Bioanalytical Method Validation Guidance. The parameters include selectivity, linearity, Lower Limit Of Quantification (LLOQ), accuracy and precision, recovery, carry over, dilution integrity, matrix effect and stability19. Therefore, this study aims to obtain a validated method for AA in dried blood spots with propranolol as its internal standard.
MATERIALS AND METHODS
This study was conducted in December, 2018 to June, 2019 at the Bioavailability and Bioequivalence Laboratory, Faculty of Pharmacy, Universitas Indonesia, Depok, West Java, Indonesia.
Materials: Acrylamide and propranolol were obtained from Sigma-Aldrich (Singapore). Formic acid, acetonitrile and methanol were a product of Merck (Darmstadt, Germany). Aqua bidestilata was purchased from Wiloso (Indonesia). Blood with hematocrit 35, 38, 40, 42, 45 and 48% were obtained from Indonesian Red Cross Society/Palang Merah Indonesia (Indonesia). The Perkin Elmer 226 (Waltham, USA) was used for analysis.
UPLC-MS/MS condition: The experiment performs on an ACQUITY™ UPLC system (Waters Corp., Milford, MA, USA) and a Xevo TQD triple quadrupole mass spectrometer (Waters Corp., Manchester, UK) equipped with positive electrospray ionization (ESI+). Data were obtained by the MasslynxTM NT4.1 software and interpreted by QuanLynxTM program (Waters Corp., Milford, MA, USA). A reverse-phase Acquity® UPLC BEH C18 column (1.7 μm, 2.1×100 mm, Waters, Milford, MA, USA) was utilized to separate the analyte. The flow rate was 0.2 mL/min under gradient condition with a combination of formic acid 0.1%-acetonitrile as the mobile phase. The injection volume was 10 μL and the duration of elution was 3 min.
Preparation of stock and working solution: The acrylamide stock solution was prepared by dissolving 10 mg of AA to 10 mL of ultrapure water. This stock solution was prepared by diluting of stock solution. Propranolol stock solution was prepared at 1.0 mg mL–1 in methanol. This stock solution diluted to obtain a working solution. All solutions were stored at 4oC and brought to room temperature before use.
Preparation of calibration standards and Quality Control (QC) samples: The calibration standards consisted of a minimum of seven concentrations, a blank sample (without IS) and zero samples (with IS). Calibration standard concentrations (2.5, 5, 7.5, 15, 50, 25, 75 and 100 μg mL–1) were obtained by spiking 500 μL blood. Four different QC samples for acrylamide were prepared separately at the concentration of 2.5, 7.5, 50 and 100 μg mL–1 for Lower Limit of Quantification (LLOQ), quality control low, medium and high.
Preparation of sample in dried blood spot: The calibration and QC sample solution were spotted to DBS cards with volume 30 μL and let dry for at least 2 h. Complete drying process was indicated by blood colors changing from red to brownish color. The circled spot is cut and transferred to the microtube. The internal standard with a concentration of 10 μg mL–1 was added to the microtube for 100 μL. Methanol as extraction solvent was added for 500 μL to the microtube. Then, the tube was being a vortex for 1 min, sonicated for 5 min and centrifugated at 4.015 g for 1 min. After that, 400 μL supernatant was evaporated under nitrogen with the 40°C and pressure 8 psi. The dried sample was reconstituted with 100 μL of 0.1% formic acid-acetonitrile (60:40) and sonicated for 20 min. Then, the sample was vortexed for 30 sec and centrifugated at 737 g for 3 min. Finally, 10 μL of sample was injected to UPLC-MS/MS .
System suitability test: System suitability was tested after optimization of analytical condition to ensure the quality performance throughout the run. The acceptable criteria for system suitability20 is CV%<6%.
Selectivity: The selectivity was estimated by investigating six blank matrices and blank matrix spiked with AA and IS. The endogenous interference co-eluted with the analyte’s peak areas should be lower than 20% of the peak area of the LLOQ standard and lower than 5% of the peak area of the IS19.
Carry-over: The carry-over parameter was valued by injecting Upper Limit of Quantification (ULOQ), blank and LLOQ, respectively for five times. The peak area should be less than 20% of the peak area of analyte at LLOQ and 5% of the peak area of IS19.
Lower Limit of Quantification (LLOQ): The lower limit of quantification was assessed by analyzing blank matrix spiked with ½ or ¼ of the acrylamide lowest concentration. This analysis was run for five replicates in at least 3 times runs. The analyte response for accuracy should be ±20% of nominal concentration and precision should be ±20% CV19.
Linearity and range: Linearity and range were obtained from the calibration curve. It includes a least six concentrations, plus blank sample (without IS) and zero sample (with IS). Linearity for biological fluid was acceptable when correlation (r) was greater than 0.98 and the differential percentage was within ±15% for nominal concentration, ±20% for LLOQ. All calibration curves were calculated by the weighted regression method (1/x) by the peak area ratios of the analyte to the internal standard versus the nominal concentration19.
Accuracy and precision: Accuracy and precision were evaluated for intra and inter-day by assaying five replicates of LLOQ, L, M and H quality control in three consecutive validation days. The criteria accepted with ±15% relative error from the theoretical values and within ±15% relative standard deviation except for at LLOQ, where it should not differ by more than19 ±20%.
Recovery: Recovery results were evaluated by dividing extracted samples by extracts of blanks spiked with the analyte post-extraction at L, M and H quality control. The criteria for coefficient variance (CV%) on recovery values should not be greater than19 ±15%.
Dilution integrity: Dilution integrity is estimated with 5 replications for each concentration. This test was carried out to overcome the sample concentration above the ULOQ calibration curve. The acceptance criteria for this test are diff% and CV% must be less than19 ±15%.
Matrix effect: The matrix effect is evaluated from six lots of blood. These blood were spiked by analyte at QCL and QCH with 2 replications for each concentration. The coefficient variance (CV%) that acceptable for this criterion must not be greater19 than 15%.
Stability: The stability of long-term and short-term stock solutions of acrylamide and propranolol were evaluated each with two replications. Long-term stability is measured on day 0 and 30. Besides, short-term stability is assessed at 0, 6 and 24 h. The diff% requirement for stock solution stability is less than ±10%19. Both long term and short term stability at low and high QC were stored inside desiccant-containing sealable plastic. The sample for short term stability was examined at 0, 6 and 24 h. The long term stability was analyzed at 0 and 28 days. The auto-sampler stability was analyzed at low and high QC with a sample left in the auto-sampler at 30oC and was measured at 0 and 24 h. The diff% provision for sample stability19 is less than ±15%.
Statistical analysis: This research does not require any statistical analysis. The full validation follows Bioanalytical Method Validation Guidance issued by FDA 2018.
RESULTS
Selection of the IS: Propranolol with the same functional group and physicochemical properties was selected to be the internal standard to increase the accuracy, precision and robustness of quantitation.
Optimization of mass condition: Both acrylamide and propranolol have basic properties due to an amine functional group in their chemical structure. Therefore, electrospray ionization in this analysis was set to positive mode. The mass spectrometric detector optimization was set with capillary voltage 3.50 kV and cone voltage 50 V. Source temperature was set to 400°C with gas desolvation flow rate 650 L h–1 and cone gas source 1 L h–1. The cone voltage for acrylamide and propranolol, respectively 26 and 36 V. The collision energy is 8 V for acrylamide and 18 V for propranolol. Figure 1 showed the fragmentation of both acrylamide and propranolol. High-intensity showed spectra signal at m/z 71.99>55.23 for acrylamide and 260.16>116.04 for propranolol.
Optimization of mobile phase combination: Three types of mobile phase combinations were tested for this optimization: (i) Formic acid 0.1%-acetonitrile, (ii) Formic acid 0.1% in water-formic acid 0.1% in acetonitrile and (iii) Methanol 5% in formic acid in 0.1% formic acid-acetonitrile. This combination was examined in isocratic methods with the composition of the aqueous phase and organic phase 50:50. The flow rate was set to 0.2 μL/min. The analyte concentration for analysis was 1 ppm. The result showed the combinations of 0.1% formic acid-acetonitrile create the best chromatogram's shape with the largest area.
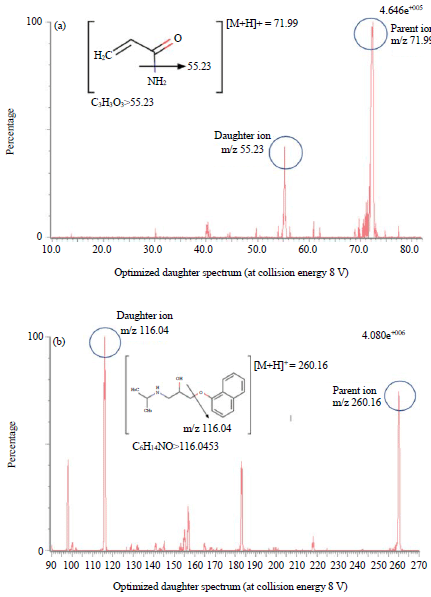 | |
| Fig. 1(a-b): | Fragmentation spectrum of (a) Acrylamide and (b) Propranolol |
| Table 1: | Gradient elution type 1 profile |
 | |
| Table 2: | Gradient elution type 2 profile |
 | |
| Table 3: | System suitability test by area |
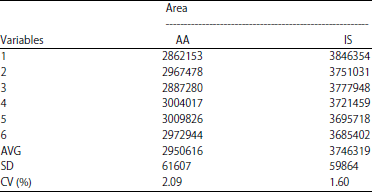 | |
| AA: Acrylamide, IS: Internal standard | |
| Table 4: | System suitability test by retention time |
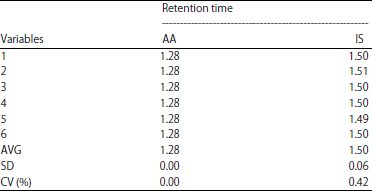 | |
| AA: Acrylamide, IS: Internal standard | |
Optimization of mobile phase composition: Composition of the optimized mobile phase with ratio 90:10, 60:40; 50:50 and 40:60 were examined for this optimization. The flow rate was set to 0.2 μL/min and concentration for analysis was 1 ppm. Based on the result, the ratio 40:60 was shown as the best composition for analysis with low retention time and the best appearance of the chromatogram.
Optimization of flow rate: The optimized combination and composition of the mobile phase were used for the optimization of the flow rate. The flow rate was tested at 0.1 and 0.2 mL/min. The chosen flow rate was 0.2 mL/min, which based on the area and the shape of the analyte chromatogram. Increasing the flow rate was necessary and faster retention time, more extensive area, with sharper peak. However, a higher flow rate increases the column pressure leads to reducing the life of the column.
Optimization of mobile phase gradient elution: Optimization under isocratic conditions provides a chromatogram with a large area, but the chromatogram presented has tailings and front lines. Therefore, gradient elution are needed to increase elution strength and create sharper peaks and larger areas. Gradient elution are shown in Table 1 and 2. These elution profiles were processed in the same duration of 3 min with changes in profile elution in 0.5, 1 and 1.2 min. The second profile was chosen based on the results which gave a larger area and better appearance of the chromatogram.
System suitability test: After optimization of analytical condition, system suitability was tested to ensure the quality performance throughout the run. System suitability test shown in Table 3 and 4. The performance of the system for both acrylamide and propranolol fulfill this criterion with CV% of area was 2.90 and 1.60%, respectively. The coefficient variance percentage of retention time for each compound was 0% and 0.24%, respectively.
Optimization of sample preparation: Several steps of this preparation were being optimized. The extraction scheme was presented in Fig. 2.
Optimization of extracting solution and volume of extracting solution: The extracting solution optimized with methanol, methanol-water (1:1) and methanol-water (4:1). The optimization uses 500 μL of solution. Based on the Peak Area Ratio (PAR), the best solution for extraction was methanol. This solution gives the most significant peak area. Then, the volume of the extracting solution was optimized. The experiment uses 500, 800 and 1100 μL of solution. The chosen volume was 500 μL. The increase of extracting volume leads to enhancement of PAR, but the duration to evaporate solution also increases, respectively. Therefore, the chosen volume for extracting the solution was 500 μL.
Optimization of vortex and sonication’s duration: Vortex is a mixing process for homogenization of heterogeneous solution. The optimization was done in the beginning and at the end of the sample preparation step with duration at 30, 60 and 180 sec. The optimal duration for each first and last vortex was at 60 and 30 sec.
 | |
| Fig. 2: | Scheme of sample preparation |
| Table 5: | Data of centrifugation rate optimization |
 | |
PAR: Peak area ratio, AA: Acrylamide, IS: Internal standard | |
| Table 6: | Data of duration of centrifugation |
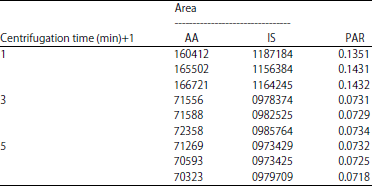 | |
| PAR: Peak area ratio, AA: Acrylamide, IS: Internal standard | |
| Table 7: | Data of acrylamide’s inter-day calibration curve |
 | |
Sonication is an ultrasound-assisted extraction. The mechanical vibration affects the molecular disruption altering the binding of sample and matrix. Beside, sonication step was optimized in the beginning and the last step of preparation. Sonication was done for 5-30 min for the first step and 5-20 min for the last step. Based on PAR, the optimum duration for first and last step sonication are 5 and 20 min for sonication, respectively.
Optimization of centrifugation’s rate and duration: Centrifugation is a critical process for sedimentation and separation of particles in solution. Clear supernatant and filtrate were needed to decrease impurities and increase column life. This optimization is tested on 16,000 and 4,000 g for 3 min. Result showed the PAR value obtained from 4000 g supernatant is greater than 16,000 g. Then, continuing to optimize the duration of centrifugation, which produces 1 min is best among 3 and 5 min centrifugation. The result of optimization of each are shown in Table 5 and 6.
All of these preparations are carried out through an enrichment process. The non-enrichment process gives different PAR results of 65.78%. Besides, optimization of the solution for recovery with methanol yields 69.89% below reconstitution with 0.1%-acetonitrile formic acid. A different reconstitution solution from an extraction solution is a factor in increasing PAR from the analyte.
Selectivity, linearity, accuracy and precision: This method is validated following FDA guidelines. Selectivity was tested in 6 different matrices in LLOQ concentration. The area of interference for this examination is 1.17-18.39% for LLOQ and 0.70-1.06% for IS. An amount of acrylamide was found in the blank sample. The minimization of this amount was done by using non-smoking subjects who have been fasting acrylamide containing foods for 14 days. The results showed a decrease in the concentration of acrylamide in the blood (<2.5 μg mL–1). Therefore, LLOQ in this study is 2.5 μg mL–1. Linearity calibration curve was observed at a concentration of 2.5-100 μg mL–1. Chromatogram peak of blank, LLOQ, ULOQ, low, medium and high QC was presented in Fig. 3. The calibration curve for 3 days has an average value of r = 0.9975. Data of inter-day calibration curve for acrylamide was shown in Table 7. Within and between run precision meet the requirements for LLOQ, low, medium and high QC. Diff% within-run for accuracy and precision about -11.73% to +17.88% for LLOQ and -2.56 to +13.71% for other concentrations with %CV for all concentrations of 12, respectively 93, 2.24, 3.5 and 4.35%.
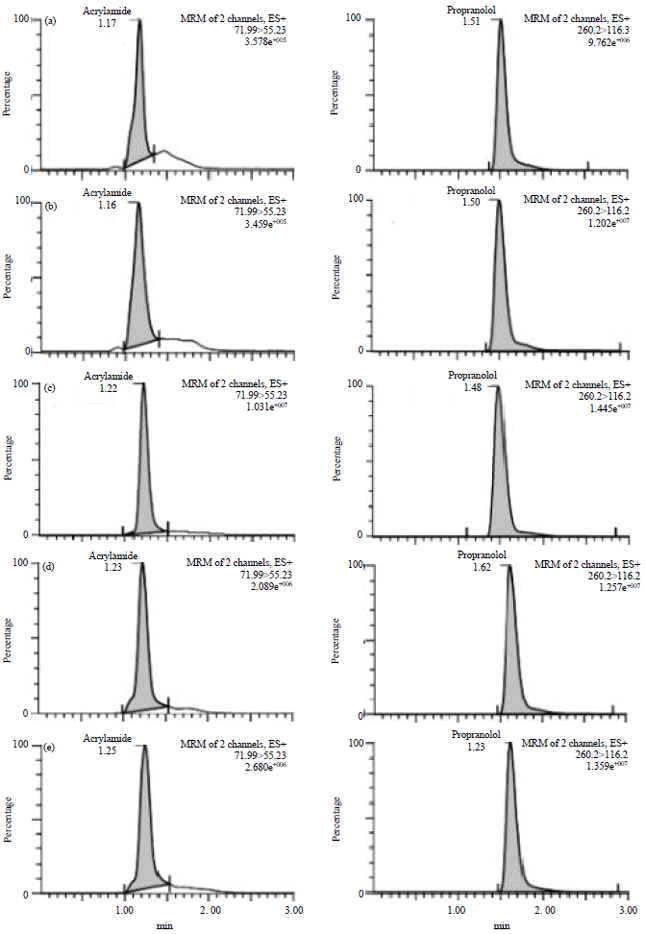 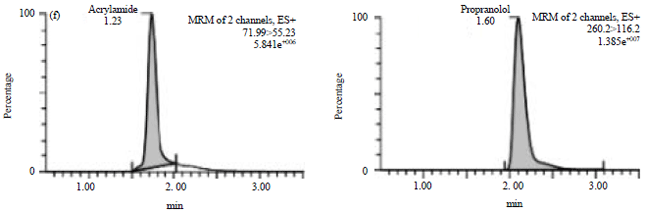 | |
| Fig. 3(a-f): | LC–ES/MS/MS MRM chromatograms (Retention time vs. Height) for acrylamide (m/z 71.99>55.23) and propranolol internal standard (260.2>116.2) in human blood (a) Blank matrices, (b) Blank matrices spiked with LLOQ concentration, (c) Blank matrices spiked with ULOQ concentration, (d) Blank matrices spiked with low QC concentration, (e) Blank matrices spiked with medium QC concentration and (f) Blank matrices spiked with high QC concentration |
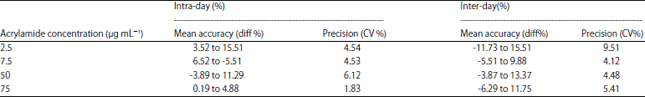
| Table 8: | Data of accuracy and precision inter and intra-day |
 | |
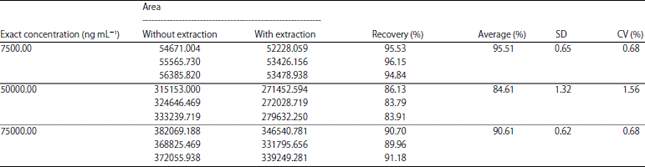
| Table 9: | Data of acrylamide recovery process |
 | |
Examinations carried out gave a different% of about -11.73 to +17.88% for LLOQ and -6.25 to +11.29% for low, medium and high QC. CV% for yield-run analysis yields 9.51, 4.12, 4.48 and 5.41%, respectively. Table 8 revealed the accuracy and precision of acrylamide at low, medium and high QC. Based on these results, these parameters had fulfilled the criteria in the FDA Guidelines.
Recovery and matrix effect: The recovery was obtained with low, medium and high QC by comparing the peak area of analyte obtained from matrices spiked with the standard solution by the peak area of analyte obtained from protein precipitation protein. The percentage of recovery results for all concentration are 95.51, 84.61 and 90.61% with the CV% 0.68, 1.56 and 0.68%. Data of recovery are shown in Table 9. Even though the recovery process was not high due to matrix effect, CV% explained this process were consistent and reproducible. Data of matrix effect are shown in Table 10. The matrix effect for each low and high QC was 105 and 96.33% with CV% 14.11 and 12.42%. The IS normalized matrix was valued at 0.91 and 0.83.
Carry over: The LLOQ for this experiment is quite high because of the amount of acrylamide found in the empty matrix. Therefore, carry-over analysis is assessed by reducing the gains obtained with blank matrices. The percentage of interference was 11.89-17.44% for analyte and 0.40-0.46% for IS. Data of carry over are shown in Table 11.
| Table 10: | Data of matrix effect |
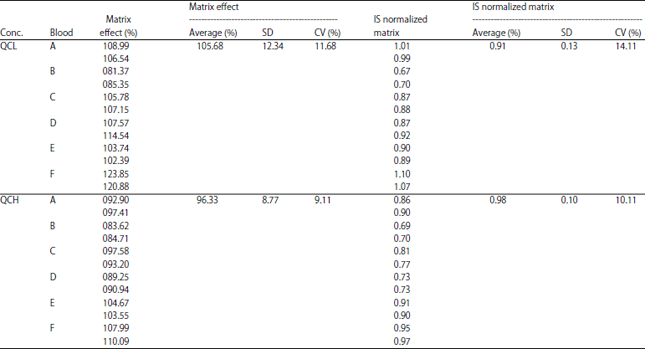 | |
| Table 11: | Data of carry over |
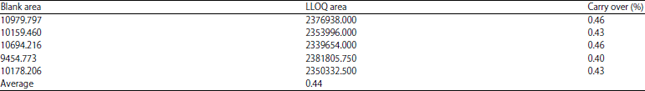 | |
| LLOQ: Lower limits of quantification | |
| Table 12: | Data of dilution integrity |
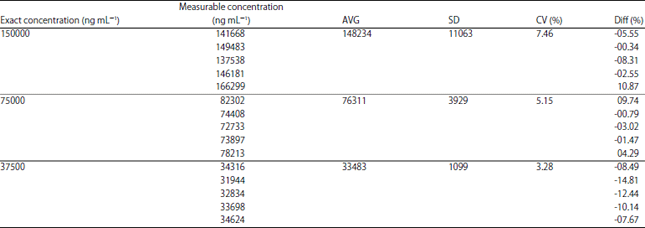 | |
| Table 13: | Data of the stability test of acrylamide (AA) |
 | |
The carry over percentage fulfills the criteria of carry over with peak area less than 20% of the peak area of analyte at LLOQ and 5% of the peak area of IS.
Dilution integrity: The concentration tested for this analysis was 37.5, 75 and 150 mg mL–1. The CV% for each of these concentrations were 3.28, 5.15 and 7.46%, with the range of diff% about -12.44-10.87%. Data of dilution integrity are shown in Table 12. The dilution integrity of AA fulfills the criteria of accuracy and precision with the CV% and diff% results below 15%.
Stability: The stability of long-term and a short-term stock solution is tested by placing stock solutions in the refrigerator (-4°C) and at room temperature (25°C) for 24 h. The stability of the stock solution gives a value of less than 10%, which means the stock solution is stable to be stored for 30 days in the refrigerator and 24 h at room temperature. Sample stability for short-term and long-term stability does not change AA concentrations in DBS cards. Data on stock solutions, sample stability and auto-sampler stability are shown in Table 13. These results indicated the sample was stable during storage and sample preparation.
DISCUSSION
In this study, the result showed the optimum condition for analyzing acrylamide was using UPLC BEH C18 and formic acid 0.1%-acetonitrile as the mobile phase with 3 min run. This method was rapid, linear in wide range concentration and has high recovery. The matrix effect for each low and high QC was 105 and 96.33% with CV% 14.11 and 12.42%. The IS normalized matrix was valued at 0.91 and 0.83. Based on the result, the matrix effect fulfills the criteria with CV%<15%19 and IS normalized matrix between 0.8-1.220.
UPLC-MS/MS usually uses an internal standard to increase the accuracy, precision and robustness of quantitation. An excellent internal standard in bioanalysis can significantly enhance the reliability of the method21. In the previous study, this assay was performed with radiolabeled isotope12,14,15. The labeled radioisotope is very expensive. Thus, compounds with the same structure and functional groups but different C-H lengths can be used to overcome the use of expensive Stable Isotope Labeled (SIL). Chemical substance which is similar to acrylamide not applicable as internal standard due to the reactivity of AA with amino, thiol and carbonyl group22. In this study, propranolol with the same functional group and physicochemical properties was selected to be the IS. The method was fully validated to demonstrate the linearity, accuracy, precision, process recovery and stability using propranolol as internal standard.
Dried blood spot bio-sampling method was used to obtain acrylamide concentration in human blood. Sample preparation in this study was using protein precipitation technique. This technique is typical for whole blood matrices, quite fast and more economist23,24.
The concentration of LLOQ in previous study which assayed plasma, urine and 14 different tissues of rat were lower than in this study12. The LLOQ valued in this study was 2.5 μg mL–1. The amount of acrylamide was quite massive due to certain acrylamides were found in the blank sample of human bloods. Acrylamide levels vary depending on the amount of acrylamide that contaminated and formed in the human body. This compound easily found in environment, food and beverage which quickly absorbed into systemic and then distributed to all tissue1-3,6,25,26. Therefore, further investigation of acrylamide in the human body must be carried out.
CONCLUSION
The specific and sensitive UPLC-MS/MS method for the determination of acrylamide in DBS was successfully developed and validated. Several acrylamides were found in the blank matrix. Therefore, the LLOQ assessed in this study was 2.5 μg mL–1. The IS, propranolol can be used for acrylamide measurement and can control the matrix effects.
SIGNIFICANCE STATEMENT
This study discovers validated method of acrylamide in dried blood spot using propranolol as internal standard that can be beneficial for analysis of acrylamide in human blood. This study will help the researcher to uncover the concentration of acrylamide substance in human blood using dried blood spot as biosampling method and propranolol as the internal standard.
ACKNOWLEDGMENT
Directorate of Research and Community Services Universitas Indonesia, Depok, Indonesia, for the financial support of this research with Grant Number: NKB-0480/UN2.R3.1/HKP.05.00/2019.
REFERENCES
- IARC., 1994. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 60, IARC Press, Lyon.
Direct Link - Saboktakin, M.R., 2014. Acrylamide, Synthesis and Properties. Nova Science Publishers, New York.
Direct Link - Bergmark, E., 1997. Hemoglobin adducts of acrylamide and acrylonitrile in laboratory workers, smokers and nonsmokers. Chem. Res. Toxicol., 10: 78-84.
CrossRefDirect Link - Papoušek, R., Z. Pataj, P. Nováková, K. Lemr and P. Barták, 2014. Determination of acrylamide and acrolein in smoke from tobacco and e-cigarettes. Chromatographia, 77: 1145-1151.
CrossRefDirect Link - Kocadağli, T. and V. Gökmen, 2015. Metabolism of Acrylamide in Humans and Biomarkers of Exposure to Acrylamide. In: Acrylamide in Food: Analysis, Content and Potential Health Effects, Gökmen, V. (Ed.)., Chapter 6. Academic Press, New York, pp: 109-128.
Direct Link - Segerbäck, D., C.J. Calleman, J.L. Schroeder, L.G. Costa and E.M. Faustman, 1995. Formation of N-7-(2-carbamoyl-2-hydroxyethyl) guanine in DNA of the mouse and the rat following intraperitoneal administration of [14C] acrylamide. Carcinogenesis, 16: 1161-1165.
CrossRefDirect Link - Settels, E., U. Bernauer, R. Palavinskas, H.S. Klaffke, U. Gundert-Remy and K.E. Appel, 2008. Human CYP2E1 mediates the formation of glycidamide from acrylamide. Arch. Toxicol., 82: 717-727.
CrossRefDirect Link - Sumner, S.C.J., T.R. Fennell, T.A. Moore, B. Chanas, F. Gonzalez and B.I. Ghanayem, 1999. Role of cytochrome P450 2E1 in the metabolism of acrylamide and acrylonitrile in mice. Chem. Res. Toxicol., 12: 1110-1116.
CrossRefDirect Link - Dearfield, K.L., G.R. Douglas, U.H. Ehling, M.M. Moore, G.A. Sega and D.J. Brusick, 1995. Acrylamide: A review of its genotoxicity and an assessment of heritable genetic risk. Mutat. Res./Fundam. Mol. Mech. Mutagen., 330: 71-99.
CrossRefDirect Link - Sega, G.A., E.E. Generoso, P.A. Brimer and H.V. Malling, 1990. Acrylamide exposure induces a delayed unscheduled DNA synthesis in germ cells of male mice that is correlated with the temporal pattern of adduct formation in testis DNA. Environ. Mol. Mutagen., 16: 137-142.
CrossRefDirect Link - Kim, T.H., S. Shin, K.B. Kim, W.S. Seo and J.C. Shin et al., 2015. Determination of acrylamide and glycidamide in various biological matrices by liquid chromatography-tandem mass spectrometry and its application to a pharmacokinetic study. Talanta, 131: 46-54.
CrossRefDirect Link - Barber, D.S., J. Hunt, R.M. LoPachin and M. Ehrich, 2001. Determination of acrylamide and glycidamide in rat plasma by reversed-phase high performance liquid chromatography. J. Chromatogr. B: Biomed. Sci. Applic., 758: 289-293.
CrossRefDirect Link - Annola, K., P. Keski-Rahkonen, K. Vähäkangas and M. Lehtonen, 2008. Simultaneous determination of acrylamide, its metabolite glycidamide and antipyrine in human placental perfusion fluid and placental tissue by liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. B, 876: 191-197.
CrossRefDirect Link - Kopp, E.K. and W. Dekant, 2009. Toxicokinetics of acrylamide in rats and humans following single oral administration of low doses. Toxicol. Applied Pharmacol., 235: 135-142.
CrossRefDirect Link - Ostler, M.W., J.H. Porter and O.M. Buxton, 2014. Dried blood spot collection of health biomarkers to maximize participation in population studies. J. Visual. Exp., Vol. 83.
CrossRefDirect Link - Sharma, A., S. Jaiswal, M. Shukla and J. Lal, 2014. Dried blood spots: Concepts, present status and future perspectives in bioanalysis. Drug Test. Anal., 6: 399-414.
CrossRefDirect Link - Giavarina, D. and G. Lippi, 2017. Blood venous sample collection: Recommendations overview and a checklist to improve quality. Clin. Biochem., 50: 568-573.
CrossRefDirect Link - Briscoe, C.J., M.R. Stiles and D.S. Hage, 2007. System suitability in bioanalytical LC/MS/MS. J. Pharm. Biomed. Anal., 44: 484-491.
CrossRefDirect Link - Zamora, R., R.M. Delgado and F.J. Hidalgo, 2015. Use of Nucleophilic Compounds, and Their Combination, for Acrylamide Removal. In: Acrylamide in Food: Analysis, Content and Potential Health Effects, Gökmen, V. (Ed.)., Academic Press, New York, pp: 297-307.
Direct Link - Burgess, R.R., 2009. Protein precipitation techniques. Methods Enzymol., 463: 331-342.
CrossRefDirect Link - Bergmark, E., C.J. Calleman, F.S. He and L.G. Costa, 1993. Determination of hemoglobin adducts in humans occupationally exposed to acrylamide. Toxicol. Applied Pharmacol., 120: 45-54.
CrossRefDirect Link - Mojska, H., 2015. Secular Trends in Food Acrylamide. In: Acrylamide in Food: Analysis, Content and Potential Health Effects, Gökmen, V., (Ed.)., Academic Press, New York, pp: 39-59.
Direct Link