
Farhina Pasha
Department of Biology, Faculty of Science, University of Tabuk, 71491 Tabuk, Kingdom of Saudi Arabia
LiveDNA: 91.32468
Aishah Alatawi
Department of Biology, Faculty of Science, University of Tabuk, 71491 Tabuk, Kingdom of Saudi Arabia
Manal Amir
Department of Biology, Faculty of Science, University of Tabuk, 71491 Tabuk, Kingdom of Saudi Arabia
Uzma Faridi
Department of Biochemistry, University of Tabuk, Tabuk Saudi Arabia
LiveDNA: 91.32541
Pakistan Journal of Biological Sciences
Year: 2020 | Volume: 23 | Issue: 8 | Page No.: 1086-1095







ABSTRACT
Background and Objective: The epidemiology of Nipah virus (NiV) was shortly seen in many Asian countries like Malaysia, Bangladesh and India most recently. Nipah virus also synonym as bat born virus is transmitted primarily by fruit bats. The 2 different strains transmitted are Hendra (highly pathogenic) and Cedar (non-pathogenic). The present study was attempt to develop recombinant protein based reagents for molecular diagnosis of Nipah. Materials and Methods: The different primer sets were developed using bioinformatics software DNASTAR. The E. coli cells were used for recombinant protein expression. Results: The NiV ‘G’ region primers were designed and amplified for 1 kb fragment and cloned. The NiV ‘G’ fragments were sub-cloned in pET-28(+) B and pGEX-5x-1. Recombinant protein thus obtained in soluble form in both the cases was essayed using western blot. The result showed the protein expression yield was more in pET-28(+) B with low stability and vice versa for pGEX-5x-1. Conclusion: The antibodies raised from the protein can be used as diagnostic reagent for detection of NiV. Thus, a new diagnostic technique can be industrialized.
PDF Abstract XML References Citation
Copyright: © 2020. This is an open access article distributed under the terms of the creative commons attribution License, which permits unrestricted use, distribution and reproduction in any medium, provided the original author and source are credited.
How to cite this article
Farhina Pasha, Aishah Alatawi, Manal Amir and Uzma Faridi, 2020. Development of Molecular Diagnosis by PCR for the Detection of Infection and Gene Expression for Nipah Virus (NiV). Pakistan Journal of Biological Sciences, 23: 1086-1095.
DOI: 10.3923/pjbs.2020.1086.1095
URL: https://scialert.net/abstract/?doi=pjbs.2020.1086.1095
DOI: 10.3923/pjbs.2020.1086.1095
URL: https://scialert.net/abstract/?doi=pjbs.2020.1086.1095
INTRODUCTION
Nipah Virus (NiV) derives its name from Kampung Sungai Nipah, a village in Malaysia from where it was first reported in 1998-1999. The virus comes under Henipavirus (a relatively new genus) in the sub-family Paramyxovirinae1. NiV is closely related to two other bat borne viruses, the Hendra virus which is highly pathogenic2 and often fatal to camels, horses and other mammals including humans3 and the Cedar virus which is generally considered non-pathogenic. NiV is a single stranded negative sense RNA virus, pleomorphic ranging from 40-600 nm in diameter covered by a lipid bilayer envelope.
The virus has three core proteins, the Phosphoproteins (P), large polymerase proteins (L) and Nucleocapsid proteins (N), The casing has two forms of spikes; Fusion proteins (F) and receptor-binding Glycoproteins (G). There are C, V, and W three additional proteins responsible for transcription and replication of the virus. Normally bats of Pteropus genus i.e., fruit eating bats are the main pool4,5, bats of some other order may also serve as a source of infection for the virus. This also include insectivorous bats. Normally the virus is circulated between bats and mammals (especially pigs in particular). Although, anti-NiV have been found in cats, dogs, goats and horses. The bats don’t pass the infection by direct contact, but propagate the virus in their secretion and excreta. If the humans come in direct contact with these fluids or excreta or partially eaten or licked fruits from these infected bats they acquire the infection. Mammals infected (pigs)6 showed respiratory and neurological symptoms. Cough is a vital sign, human to human spread of the virus has been recorded in health care providers7. Although, it is not known clearly as to how long the virus survives in hospital environment, but its persistence is of great concern. As a fact there is limited records for NiV being present in body fluids, partially eaten bat fruits and in date palm sap8. The incubation period can range from as short as 2 days up to 45 days maximum. Humans might be infected all through this period. The primary symptoms are in humans are similar to flu such as fever and headache etc. but acute encephalitis may soon develop which may result in mortality. Those survive could later experience neurological, personality disorders and seizures. Most of the laboratories depend on techniques such as immuno fluorescent. RT-PCR and ELISA. There is a demand for rapid detection as well as, serological diagnosis of virus for monitoring the presence of virus and its antibodies in individual and animals in high-risk areas. Currently production of immunological reagents for their assay requires Biosafety level-4 laboratories, which are limited only to a few countries worldwide. At the Institute of Bioscience, Malaysia, the ‘G’ protein of NiV had been cloned and expressed in E. coli and used as capturing reagent in ELISA to determine the presence of anti NiV antibodies in serum samples collected from naturally infected swine9-12. A recombinant subunit vaccine has been recently formulated against lethal NiV challenge in cats13. One vaccine named as ALVAC, Canarypox vectored Nipah F and G was said to be a potential for swine as well as human9,14. Considering the situation15, there is an urgent need to undertake research. The present study was aimed at optimization of molecular diagnosis by PCR, which is based on the amplification of specific region of the genome which is accurate and preferred choice of diagnosis in recent times and to develop recombinant protein based reagents in the laboratory.
MATERIALS AND METHODS
Study area: The study was carried out in Laboratory of Biology Department, University of Tabuk, Saudi Arabia, from September, 2017-June, 2019 for nonpathogenic strain. All the chemicals used were analytical grade. Media-bacteriological media used in the study was prepared according to standard procedures.
Standard clone: The cloned NiV ‘G’ gene was generously obtained from Malaysia for nonpathogenic strain (Sino Biological). Immunoconjugates-Penta-Histidine-Horseradish-peroxidase-conjugate (Novagen Pharma) and Glutathione-sulfonyl transferase Horseradish-peroxidase conjugate (Pharmacia) were used in this study.
Oligonucleotides: The sequences of oligonucleotides were in a stock of 50 n mole and further diluted to stock of 10 p mole/reaction (Sigma@life sciences). The different primer sequences along with their respective ID are presented in Table 1.
Sampling: A total of 165 clinical samples including swine spleen, liver, brain, lung, blood and bat fecal samples were collected. All sample processed were for nonpathogenic strain.
| Table 1: | Primer sequences with respective ID |
 | |
RNA isolation: The RNA isolation was performed as outlined in the user manual of the QIAgen RNA Purification Kit (QIAgen, Saudi Arabia).
Different sets of primers designed with the help of bioinformatics software DNA STAR (Lasergene) were used to amplify various fragments of ‘G’ gene of NiV in the standard clone. The Polymerase Chain Reactions (PCR) was performed in Gene Amp PCR (Labnet, Multigene optimax, Saudi Arabia) as described by Sambrook et al.16 with slight modifications.
Agarose gel electrophoresis: Agarose gel electrophoresis was performed in gel electrophoresis system as described by Sambrook et al.16.
Gel extraction: Gel extraction was done as recommended in the instruction manual of QIAgen's gel extraction kit (QIAgen, Saudi Arabia). The concentration of the eluted product was checked with agarose gel electrophoresis.
Cloning of the PCR amplified DNA segments of ‘G’ gene of Niv was done in pGEM-T Easy Vector (Promega Fig. 1). The insert was then released from pGEM-T Easy. Later sub-cloned in two expression vector systems i.e., pGEX-5X-1 (Pharmacia, Fig. 2) and pET 28 b (Novagen, Fig. 3) for expression ‘G’ gene in prokaryotic (bacterial) cells.
Competent cells: were prepared by Calcium Chloride method as described by Sambrook et al16.
Recombinant selection colony selection was done by following assays as described by Sambrook et al.16. The colonies with non-recombinant plasmids gave blue colonies while with the recombinant plasmids white colonies were produced. The white colonies were selected for mobility shift assay. The suspected colonies were individually streaked on ampicillin plate. Recombinant plasmid isolation was done using the Minipreps DNA Purification system. The purity and yield of the extracted plasmid DNA were checked in 1% agarose gel by electrophoresis.
Restriction digestion: To confirm the presence of insert in recombinant plasmids, restriction digestion was done with compatible enzymes i.e., restriction digestion with Not-I enzyme. To check the orientation of insert in cloning vector orientation PCR was done. The PCR cycling conditions were the same as mentioned in PCR section above.
• Nucleotide sequencing: Sanger’s dideoxy method was followed for nucleotide sequencing2 and was carried out in a semi-automatic sequencer
• Autoradiograph: was established by exposing the gel to X-ray film and later developed. Both the vectors were checked in 1% agarose gel for linearization and then were gel extracted. Again the yield and purity was checked in 1% agarose gel
• Protein extraction: Plasmid was isolated and retransformed in BL-21 DE3 cells and in BL-21 cells using CaCl2 method. Further plating was done on LB agar media containing respective antibiotics
 | |
| Fig. 1: | Cloning vector pGEM-T easy |
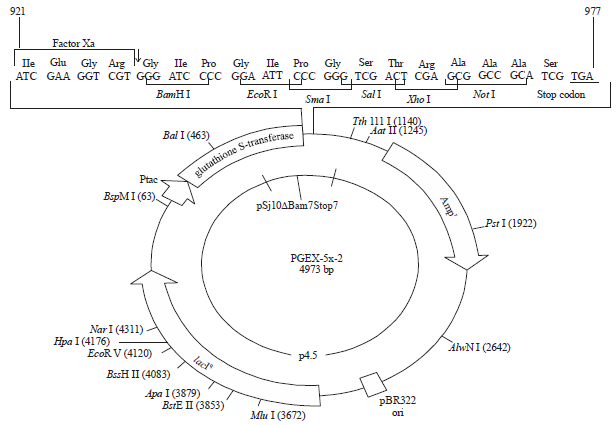 | |
| Fig. 2: | Expression vector pGEX-5x-1 |
 | |
| Fig. 3: | Expression vector pET 28 b(+) |
Recombinant selection: Was done using colony PCR. Recombinant plasmid isolation was done using Wizard Plus SV Minipreps DNA purification system. The purity and yield of extracted plasmid was checked in 1% agarose gel electrophoresis.
• Restriction digestion: It confirmed the presence of insert in recombinant plasmids. Orientation of ‘G’ gene in both the vectors was confirmed by orientation PCR amplification
• Protein extraction: It was done using manual provided with big buster protein extraction reagent (Novagen). The clarified extracts were maintained on ice for short-term storage (2-3 h) and further frozen at -20°C until required
Confirmation of gene expression: The induced and non-induced pellet were analyzed by sodium dodecyl sulphate-poly acrylamide gel electrophoresis (SDS-PAGE). Two separate gels were cast in the same apparatus opposite to each other one for staining and other for western blotting, protein bands was visualized.
RESULTS
Optimization of PCR: Optimization of PCR for the detection of NiV infection was done using a standard clone of ‘G’ gene. These primers were found to be specific and yielded amplicon of specific size Fig. 4. Also, 1 kb fragment of G gene was amplified using same primers sets in different combinations and was visualized on 1% agarose gel (Fig. 5). A total of 165 clinical samples of swine spleen, liver, brain, lung, blood and bat fecal samples were processed for RNA isolation. By using the reference ‘G’ clone of NiV, the field samples were screened for NiV infection (targeted region G of NiV), which were all found to be negative in conventional PCR.
Cloning of amplicon: Amplified products (1 kb fragment of G gene) were resolved in 1% agarose gel which was excised and the DNA was extracted, the yield and purity of the extracted product were found to be satisfactory. Cloning of the amplified segments of G gene of NiV was done in pGEM-T Easy Vector. The recombinant colonies obtained on LB-AIX plates were screened by blue white selection (Fig. 6).
Confirmation of clones: Mobility shift assay of super coiled plasmid was performed and 4 kb product was seen on 1% agarose gel (vector size 3 kb and insert size 1 kb, Fig. 7). The plasmids were extracted from the recombinant colonies and the yield and purity of the extracted plasmids, checked by 1% agarose gel electrophoresis were found to be satisfactory.
 | |
| Fig. 4: | Amplification of various fragments of NiV ‘G’ gene in standard clone 6532 |
Lane M-100 bp DNA ladder, Lane 1: Non amplified plasmid , Lane 2: 473 bp amplicon, Lane 3: 440 bp amplicon, Lane 4: 380 bp amplicon, Lane 5: 396 bp amplicon | |
 | |
| Fig. 5: | Amplification of 1 kb fragment of NiV ‘G’ gene, Lane M- 100 bp DNA ladder |
| Lane 1, 2, 3, 4, 5, 6- clone no.6532 yielding 1 kb amplicon | |
 | |
| Fig. 6: | Blue white colony selection |
 | |
| Fig. 7: | Relative mobility shift assay of super coiled plasmid, Lane M-super coiled DNA ladder from 2 kb onwards |
| Lane 1-3: Recombinant plasmid with insert of 4 kb | |
 | |
| Fig. 8: | Release of insert from pGEM–T Easy vector, Lane M-1kb DNA ladder |
| Lane 1, 2: Clone no 6532 releasing 1 kb insert | |
 | |
| Fig. 9: | Confirmation of orientation in pGEM-T easy vector, lane M-1kbDNA ladder |
Lane 1, 2: No amplification, Lane 3: Clone 6532 yielding 1 kb amplicon in forward orientation | |
 | |
| Fig. 10: | Sequencing autoradiography of clone 6532 |
 | |
| Fig. 11: | Colony PCR IN pET 28 (b) and pGEX-5X-1, lane M-100 bp DNA ladder |
Lane 1: Amplicon of 473 bp (clone 6533), Lane 2: Amplicon of 473 bp (clone 6535) | |
The plasmids were then subjected to restriction endonuclease analysis using Not I enzyme for the release of insert from the vector.
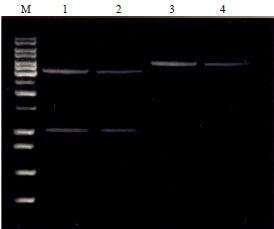 | |
| Fig. 12: | Release of insert from pET-28+(b) and pGEX-5X-1, Lane M-1kb DNA ladder |
Lane 1: Clone No. 6533 releasing 1 kb insert, Lane 2: Clone No. 6535 releasing 1 kb insert, Lane 3, 4: Clone No. 6533&6535 without releasing the insert | |
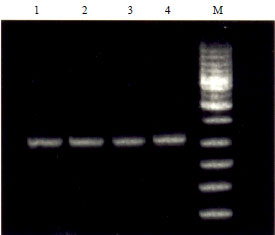 | |
| Fig. 13: | Confirmation of orientation in pET-28+(b) and pGEX-5X-1, lane M-1kb DNA ladder |
Lane 1, 2: Clone No. 6533 yielding 1 kb amplicon, Lane 3, 4: Clone No. 6535 yielding 1 kb amplicon | |
The specific sizes insert (1 kb) was released and visualized on 1% agarose gel electrophoresis (Fig. 8). Orientation PCR using M13 F primer and NiVBDG-31R confirmed orientation of insert in the vector. Amplicon of 1 kb was seen in 1% agarose gel (Fig. 9).
Nucleotide sequencing: The amplicon of desired size was obtained with specific primers. The autoradiograph forms 4 separate lines, one each for fragment ending with one of the four bases: A, T, C and G. The sequences were read from the autoradiograph developed (Fig. 10).
Sub-cloning of the insert in expression vector: The 1 kb insert was released from pGEM T easy with Not I enzyme and visualized on 1% agarose gel electrophoresis.
 | |
| Fig. 14: | Analysis of expressed protein by SDS PAGE in pGEX-5X-1, Lane M-Pre-stained page ruler protein marker |
Lane 1: BL21-DE3 cells control, Lane 2: Total cell pellet, Lane 3: Soluble fraction, Lane 4: Inclusion bodies | |
 | |
| Fig. 15: | Analysis of expressed protein in Pgex-5X-1 by SDS PAGE, Lane M-Page ruler pre-stained protein marker |
Lane 1: BL21 cells (control), Lane 2: Total cell pellet, Lane 3: Soluble fraction, Lane 4: Inclusion bodies | |
The band of specific size (1000 bp) was than gel extracted and subcloned in two different expression vector systems i.e., pET 28 (+) b (novagen) and pGEx-5x-1 (Pharmacia) (both of which were linearized with Not I) and transformed in E. coli JM109 cells. The colonies obtained were screened by colony PCR, orientation PCR and RE analysis as stated below.
 | |
| Fig. 16: | Western blot analysis of expressed protein in pET-28-(b), Lane M-Page ruler pre-stained protein marker |
| Lane 1: BL21 cells (control), Lane 2: Total cell pellet, Lane 3: Soluble fraction, Lane 4: Inclusion bodies | |
 | |
| Fig. 17: | Western blot analysis of expressed protein in pGEX-5X-1, Lane M-Page ruler pre-stained protein marker |
| Lane 2: BL21 cells (control), Lane 3: Soluble fraction, Lane 4: Inclusion bodies | |
Restriction enzyme analysis: A single isolated colony from LB Kan plate (for pET vector which has a kanamycin resistance) and from LB amp plates (for pGEx which has ampicillin resistance) were subjected to colony PCR and product of 1 kb was seen on 1% agarose gel (Fig. 11). Forward orientation of insert in pET 28 (+) b was confirmed by orientation PCR using T7 reverse and NiVBDG-28F primer. Similarly amplified product of 1 kb was seen when orientation of insert in pGEx-5x-1 was checked using pGEx upstream primer and NiVBDG 31R, thus confirming the forward orientation of insert Fig. 12. Colonies found positive by above assays were subjected to plasmid isolation (for both the vector systems). The isolated plasmids which were than subjected to restriction endonuclease analysis using Not I enzyme released the insert of specific size (1 kb) which was visualized on 1% agarose gel (Fig. 13).
Plasmid extraction: The plasmids extracted from colonies on LB kan plates and found positive by above assays were retransformed in BL-21 DE3 cells (expression hosts for pET vector). Also, plasmids extracted from colonies on LB amp plates and found positive were retransformed in BL-21 cells (expression host for pGEx). Single isolated colony from each plate (LB amp and LB kan ) was used for induction. The expression of the ‘G’ protein in both the vector systems was carried at second passage level cultures, at log phase growth, with an optimum optical density of 0.6 and inducing the cultures with 1 mM final concentration of IPTG for 4 h. The cells were pelleted after 4 h induction and inclusion body and soluble fraction from the cell pellet along with non-induced culture (control) were subjected to SDS-PAGE analysis using prestained protein marker as standard (Fig. 14 and 15). After staining the gel a prominent high intensity band in soluble fraction at 37 kDa was observed in case of pET 28(+) b and a prominent low intensity band in soluble fraction at 63 kDa (in case of pGEx-5x-1 since actual protein size is 37 kDa and size of GST tag is 26 kDA) was observed in induced cultures, which was absent in non-induced control indicating the expressed protein.
Protein expression: The expression of protein in pET 28(+) b was confirmed by western blotting using Penta-His HRP conjugate (Novagen). The expression of protein in pGEx-5x-1 was also confirmed by western blotting using GST-HRP conjugate (Fig. 16 and 17).
DISCUSSION
The virus in family Paramyxoviridae, the cell attachment protein of NiV lacks both Haemagglutination and neuraminidase activity and hence, are designated as ‘G’ proteins14. This ‘G’ protein plays a central role in the viral replication processes and is responsible for virus attachment to sialic acid containing host cell receptor4. Therefore, the ‘G’ protein of NiV is considered as primary target for neutralizing antibodies6. All the clinical samples viz.tonsils, brain, lung, spleen, and whole blood from apparently healthy swine as well as randomly selected bat fecal3 did not yield any positive reaction against targeted ‘G’ region of NiV by PCR (nonpathogenic strains). A clone of ‘G’ gene of NiV was obtained as a reference material with the help of which optimization of PCR for molecular diagnosis of NiV infection was done using region specific primers. Instead of using this expression method a portion of extracted ‘G’ gene was cloned in pGEM T Easy vector. From these recombinant colonies the plasmid was extracted the yield and purity of which was found to be satisfactory when checked on 1% agarose gel electrophoresis. The plasmids extracted from recombinant colonies had satisfactory purity. Specific vector band at 3 kb and insert band at 1 kb was visualized on 1% agarose. Also, orientation PCR confirmed the forward orientation of insert in the vector. A single isolated colony was subjected to PCR and 1 kb product was detected, further it was confirmed to be in forward orientation by orientation PCR. Thus, NiV infection can be detected by RT-PCR with high accuracy, sensitivity and repeatability if specific primers are used. The recombinant ‘G’ protein could be expressed in BL-21 and BL21-DE3 cells. The intensity of protein band in the SDS-PAGE gel was more in case of pET 28 b while a faint band was observed in case of pGEX-5x-1 thus indicating higher expression levels of protein in pET vector system. In spite of high levels of expression in pET-28-b the stability of the protein was very less i.e., the protein could be expressed only to second passage level culture. On the contrary in case of pGEX-5x-1 the protein was stable up to third passage level inspite of poor yield. The expression of ‘G’ proteins of NiV in baculovirus expression system ranges between 100-200 μg 10–6 cells17. The yield of purified truncated ‘G’ protein reported18 was also in similar range. The very poor yield in the present study could probably be due to prokaryotic expression systems used. The potential diagnostic utility of recombinant ‘G’ protein has been explored for nonpathogenic strain, which facilitates the detection of NiV infection. It has an advantage that it eliminates the necessity of use of infectious virus in diagnostic tests. Nonetheless more studies are needed to access the use of recombinant ‘G’ protein in routine diagnosis for positive strains since it requires standardization of ELISA by using known Nipah positive sera and level 4 biosafty lab. In May, 2018 the recent NiV epidemic occurred in two northern districts of Kerala, India. Total of 16 mortal cases were reported. The main center of the epidemic was Kozhikode located in Malappuram district. The occurrence of the NiV was confirmed by RT-PCR. Although, the initial test did not confirm virus in bats, but the later confirmed (News minutes; Ministry of Health, India) thereby establishing transfer of infection via bats. A new case was reported on 4 June, 2019 from Kochi India (News, Mathrubhumi, Retrieved 6, June). Previously also bats have been observed as main reservoir for NiV transmittance from India and Bangladesh,2,13,17-20 whereas, pigs as main reservoir in Malaysia and Singapore. Therefore, research in virology must be given priority as it has an immense potential of becoming pandemic at a very fast pace. Therefore, research for viruses like Nepah, Zika, Ebola should be on priority. The hope lies in the proper deciphering of host -parasite interaction thus obtaining sufficient information for the development of antiviral vaccines and drugs.
CONCLUSION
In the present study NiV gene was successfully amplified. Its expression was of significant yield. The recombinant protein obtained was in soluble form therefore can serve as important laboratory diagnosis tool. Potential diagnostic utility of recombinant ‘G’ protein has been established which can be effectively used for detection of Nipah virus infection.
SIGNIFICANCE STATEMENT
This study aims to develop novel molecular diagnosis for Nipah virus. As the basis of this molecular diagnosis is a specific gene amplified from genome, the result accuracy is very high. The technique promises a new diagnosis tool for NiV epidemic.
REFERENCES
- Wang, L.F., W.P. Michalski, M. Yu, L.I. Pritchard, G. Crameri, B. Shiell and B.T. Eaton, 1998. A novel P/V/C gene in a new member of the paramyxoviridae family, which causes lethal infection in humans, horses and other animals. J. Virol., 72: 1482-1490.
Direct Link - Daniels, P., T. Ksiazek and B.T. Eaton, 2001. Laboratory diagnosis of Nipahand Hendra virus infections. Microbes Infect., 3: 289-295.
CrossRefPubMedDirect Link - Parashar, U.D., L.M. Sunn, F. Ong, A.W. Mounts and M.T. Arif et al., 2000. Case-control study of risk factors for human infection with a new zoonotic paramyxovirus, Nipah virus, during a 1998-1999 outbreak of severe encephalitis in Malaysia. J. Infect. Dis., 181: 1755-1759.
CrossRefPubMedDirect Link - Halpin, K., P.L. Young, H.E. Field and J.S. Mackenzie, 2000. Isolation of Hendra virus from pteropid bats: A natural reservoir of Hendra virus. J. Gen. Virol., 81: 1927-1932.
CrossRefPubMedDirect Link - Wacharapluesadee, S., B. Lumlertdacha, K. Boongird, S. Wanghongsa and L. Chanhome et al., 2005. Bat Nipah virus, Thailand. Emerg. Infect. Dis., 11: 1949-1951.
CrossRefPubMedDirect Link - Harcourt, B.H., A. Tamin, T.G. Ksiazek, P.E. Rollin, L.J. Anderson, W.J. Bellini and P.A. Rota, 2000. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology, 271: 334-349.
CrossRefDirect Link - Chadha, M.S., J.A. Comer, L. Lowe, P.A. Rota and P.E. Rollin et al., 2006. Nipah virus-associated encephalitis outbreak, Siliguri, India. Emerg. Infect. Dis., 12: 235-240.
CrossRefPubMedDirect Link - Chua, K.B., C.L. Koh, P.S. Hooi, K.F. Wee and J.H. Khong et al., 2002. Isolation of Nipah virus from Malaysian Island flying-foxes. Microb. Infect., 4: 145-151.
CrossRefDirect Link - Chua, K.B., 2003. Nipah virus outbreak in Malaysia. J. Clin. Virol., 26: 265-275.
CrossRefDirect Link - Lam, S.K. and K.B. Chua, 2002. Nipah virus encephalitis outbreak in Malaysia. Clin. Infect. Dis., 34: S48-S51.
CrossRefDirect Link - Nor, M.M., C.H. Gan and B.L. Ong, 2000. Nipah virus infection of pigs in peninsular Malaysia. Rev. Sci. Tech. Off. Int. Epiz., 19: 160-165.
Direct Link - Chua, K.B., 2003. A novel approach for collecting samples from fruit bats for isolation of infectious agents. Microbes Infect., 5: 487-490.
CrossRefDirect Link - Guillaume, V., H. Contamin, P. Loth, M.C. Georges-Courbot and A. Lefeuvre et al., 2004. Nipah virus: Vaccination and passive protection studies in a hamster model. J. Virol., 78: 834-840.
CrossRefDirect Link - Kumar, S., 2003. Inadequate research facilities fail to tackle mystery disease. Br. Med. J., Vol. 326, No. 7379.
CrossRefDirect Link - Hsu, V.P., M.J. Hossain, U.D. Parashar, M.M. Ali and T.G. Ksiazek et al., 2004. Nipah virus encephalitis reemergence, Bangladesh. Emerg. Infect. Dis., 10: 2082-2087.
CrossRefPubMedDirect Link - Joshi, R.M., 2018. Nipah virus: A novel emerging zoonotic disease. Clin. Microbiol., Vol. 7, No. 3.
CrossRefDirect Link - Yu, M., E. Hansson, B. Shiell, W. Michalski, B.T. Eaton and L.F. Wang, 1998. Sequence analysis of the Hendra virus nucleoprotein gene: Comparison with other members of the subfamily Paramyxovirinae. J. Gen. Virol., 79: 1775-1780.
CrossRefDirect Link - Wong, K.T., I. Grosjean, C. Brisson, B. Blanquier and M. Fevre-Montange et al., 2003. A golden hamster model for human acute Nipah virus infection. Am. J. Pathol., 163: 2127-2137.
CrossRefDirect Link