
A. A. Parikesit
Department of Chemistry, Faculty of Mathematics and Science, University of Indonesia, 16424, Depok, Indonesia
Kinanty
Department of Chemistry, Faculty of Mathematics and Science, University of Indonesia, 16424, Depok, Indonesia
U. S.F. Tambunan
Department of Chemistry, Faculty of Mathematics and Science, University of Indonesia, 16424, Depok, Indonesia
Pakistan Journal of Biological Sciences
Year: 2013 | Volume: 16 | Issue: 24 | Page No.: 1836-1848







ABSTRACT
Dengue virus (DENV) has spread throughout the world, especially in tropical climates. Effective treatment of DENV infection is not yet available although several candidate vaccines have been developed. Treatment at this time is only to reduce symptoms and reduce the risk of death. Therefore, antiviral treatment is much needed. Envelope protein is one of the structural proteins of DENV which is known and could be a target of antiviral inhibitors and plays a special role in the fusion process. The aim of this research is to screen the commercial cyclic peptides which are used as inhibitors of envelope protein DENV through molecular docking and molecular dynamics at 310 and 312 K. Screening of commercial cyclic peptides through molecular docking ligands obtained best 10 ligands then examined the interaction between hydrogen bonding and residue contacts of the cavity envelope protein and obtained best three ligands which could enter the cavity of envelope protein overall. The three ligands were predicted through the ADME-Tox and the best ligand obtained was BNP (7-32), porcine. The results of molecular dynamics simulations at 310 and 312 K revealed that ligand can maintain interaction with the cavity of the target.
PDF Abstract XML References Citation
Received: March 22, 2013;
Accepted: April 15, 2013;
Published: September 16, 2013
How to cite this article
A. A. Parikesit, Kinanty and U. S.F. Tambunan, 2013. Screening of Commercial Cyclic Peptides as Inhibitor Envelope Protein Dengue
Virus (DENV) Through Molecular Docking and Molecular Dynamics. Pakistan Journal of Biological Sciences, 16: 1836-1848.
DOI: 10.3923/pjbs.2013.1836.1848
URL: https://scialert.net/abstract/?doi=pjbs.2013.1836.1848
DOI: 10.3923/pjbs.2013.1836.1848
URL: https://scialert.net/abstract/?doi=pjbs.2013.1836.1848
INTRODUCTION
Dengue fever is caused by infection of Dengue virus (DENV), which is one of the dangerous pathogens faced in the world and can represent a global pandemic (Tomlinson et al., 2009). The World Health Organization estimates that 100 million cases occur each year and as many as 2.5 billion people or 40% of the world's population are at risk of being infected with DENV (Lee et al., 2007).
DENV is a virus of the genus Flavivirus, family Flaviviridae and consisted of four serotype (Farias et al., 2013). The four serotypes are DENV-1, DENV-2, DENV-3 and DENV-4. The classification is based on the type of antibody produced in the human body after infection. All four serotypes have the same morphology and genome but show different antigens so that a person can be infected with this virus more than once due to lack of complete cross protection (Lindenbach et al., 2007).
DENV contains three structural proteins; they are protein capsid (C), membrane proteins (prM) and the envelope protein (E) (Samsa et al., 2009). The entry of enveloped viruses into the host cell begins with the attachment of the virus to the receptors followed by fusion of viral envelope with the cell membrane (Teissier et al., 2011). Viral particles penetrated into the cell by endocytosis and decreasing acidity in the endosom which triggers conformational changes of DENV envelope protein dimers into monomers, followed by changes to homotrimer (Teissier et al., 2011; Kielian et al., 2010). During the transition from dimer to trimer, residues 83-100 acts as a hinge region that allows the rotation and movement of the protein E (Kielian et al., 2010). As a result of these changes, "fusion peptide" interact with the target membrane, initiating the fusion process and led to the incorporation of both host and viral cell bilayer (Dubey et al., 2011; Yennamalli et al., 2009). Various antiviral have been studied extensively and some that have been reported include viral RNA synthesis inhibitors, protein inhibitors of NS3 helicase and protease inhibitors that inhibit the maturation of DENV and polianion monoclonal antibody that prevents binding to host cell receptors (Noble et al., 2010). Research on cyclic peptides as antiviral disulfide DENV also has been done (Tambunan and Alamudi, 2010). Some targets of antiviral drugs is necessary to inhibit enzyme activity in the DENV life cycle such as protease, helicase and RNA polymerase (Sampath and Padmanabhan, 2009).
The method used in this experiment is molecular docking, as a means to predict the formation of peptide bonds between the inhibitor to be developed with the DENV envelope protein cavity. Moreover, molecular dynamics were performed to examine the interaction between proteins and cyclic peptides temperature and time. Previous research has shown that the design of the cyclic peptides as inhibitors via disulfide bridge is considered good enough to inhibit the activity of DENV protein (Tambunan et al., 2011a, b). Therefore, the screening will be focused to the developed cyclic peptides that have been sold in the market as an inhibitor of DENV envelope protein. The objective of this study was to see if commercial cyclic peptides can be used as inhibitors of DENV envelope protein during molecular docking and molecular dynamics simulation. The purpose of this study was to screen commercial cyclic peptides that can be used as an inhibitor of DENV envelope protein through the method of molecular docking and molecular dynamic.
MATERIALS AND METHODS
DENV envelope protein sequences and multiple sequence alignment: The search for the data of DENV envelope protein sequences in the human host, was done through the server at the NCBI site at the address: http://ncbi.nlm.nih.gov. DENV envelope protein sequences of the human host in FASTA format was included as input to the server ClustalW2, at the address http://www.ebi.ac.uk/Tools/clustalw2. The alignment is performed on the server against any DENV envelope protein. Envelope protein sequence with the highest score will be used as input to subsequent analysis.
Search of 3D envelope protein structure and the PDB data: The search of the 3D structure of the envelope protein was done by software Protein Model Portal (http://www.proteinmodelportal.org) provided from the multiple sequence alignment. 3D structure of the envelope protein can be downloaded from the PDB database contained in the Research Collaboratory for Structural Bioinformatics Protein Data Bank through http://www.rscb.org/pdb address.
Geometry optimization and energy minimization of the envelope protein: Before the geometry optimization and energy minimization using software MOE2008.10, the water molecules and metal molecules on the DENV envelope protein needs to be omitted. The next steps is to include the protonation of protein by selecting protonate 3D option, the addition of hydrogen atoms in the protein structure and the settings of fixed hydrogen using partial charge option. The parameters used are AMBER99 forcefield (Chen and Pappu, 2007). Energy minimization is then performed with the AMBER99 forcefield and solvation of the gas phase, as well as fixed charge carried by the RMS gradient 0.05 kcal/mol D. Other parameters are using the standard default.
Search of cyclical peptide ligands and the design of their 3D structure: The search of the cyclic peptide ligands was done through the database of the chemical company like bachem, Mimotopes and American Peptide. Peptide ligands to be used as an inhibitor of the protein envelope was drawn using ChemSketch ACDLabs (Blondelle and Lohner, 2010). Peptides are made from a combination of the three polar amino acids each end attached to the amino acid cysteine to form disulfide bridges. Ligands are drawn then converted into 3D shapes through VegaZZ software.
Geometry optimization and energy minimization of the ligand 3D structures: Ligand geometry optimization process begins by importing all the ligands into a database viewer of MOE 2008.10 and then do the wash. The goal is to improve the structure of the ligand and the position of hydrogen atoms that are present in the ligand. Furthermore, the optimization of peptide ligands is performed using AMBER99 forcefield and solvation of the gas phase. Repair of hydrogen atoms and fixed hydrogen by partial charge is also performed. Peptide ligand energy minimization was done by RMS gradient 0.001 kcal/A (Homeyer and Gohlke, 2013).
Protein ligand docking with envelope: Docking process was started by using MOE 2008.10. The selected option for simulation-dock placement and setting method used is the triangle with 100 rounds matcher. Scoring function is used to display 100 best data of London dG. Furthermore, of the 100 best displayed data, the repeated measurements (refinement) was performed based on forcefield parameters. The displayed overall results of the selected docking process is the best data, in accordance with the default parameters of the software MOE 2008.10 (Vilar et al., 2008; Tambunan et al., 2010; Majeux et al., 2001).
Determination of protein-ligand complexes conformation docking results: The results of docking calculations were shown in the output in notepad format. Determination of protein-ligand conformation docking results was done by selecting ligand conformations with the lowest binding energy.
Energy association and the inhibition constant (Ki): Bond energies and inhibition constants docking results can be seen in the output of notepad format. The protein-ligand complex was selected, it has the smallest value of binding energy and inhibition constants to do further analysis.
Hydrogen bonding and contact residues: The best results of the hydrogen bonding and contact residues of protein-ligand complexes docking results was identified by using software MOE2008.10.
ADME-tox prediction of the cyclical peptide ligands: Prediction of ADME-Tox properties (absorption, distribution, metabolism, excretion and toxicity) of disulfide cyclic peptide ligands were done online using the ACD I-labs/Percepta that was accessed through the site of https://ilab.acdlabs.com/iLab2/index.php and by offline means through Toxtree-v2.5.0 (Benigni et al., 2008).
Molecular dynamics simulation: Preparation of protein-ligand complex was required before performing molecular dynamics simulations. Geometry optimization and energy minimization was done by using the software MOE 2008.10. Setting partial charge of protein complexes-ligand was done with current forcefield parameters. Then the system uses the setting of the solvation energy minimization born and done with RMS gradient 0.05 kcal / mol D (Karplus and McCammon, 2002; Shu et al., 2011; Adcock and McCammon, 2006).
Initialization timing: The molecular dynamics simulations was performed for protein-ligand complexes at 100 ps to determine the initialization before running the main simulation.
Molecular dynamics simulation on temperature 310 K: The molecular dynamics simulations was performed for protein-ligand complexes with a temperature of 310 K with a time of major simulation during the 5000 ps and cooling for 10 ps to a temperature up to 1 K. The results of position, velocity and acceleration were saved every 0.5 ps. The other parameters were in accordance with the default-dynamic MOE.
Molecular dynamics simulation on temperature 312 K: Performed molecular dynamics simulations for protein-ligand complexes at a temperature of 312 K with heating time during 10 ps, was the main simulation during the 5000 ps and cooling for 10 ps to a temperature up to 1 K. The results of the position, velocity and acceleration are saved every 0.5 ps. The other parameters were in accordance with the default of the dynamic MOE.
Analysis of molecular dynamics simulation data: The results of molecular dynamics simulation can be seen in the output database of the viewer MOE2008.10. Interactions between protein ligands for molecular dynamics process can be viewed using LigX-Interaction.
RESULTS
Search of DENV envelope protein sequence data and multiple sequence alignment: Envelope protein sequences were obtained from the NCBI database (http://www.ncbi.nlm.nih.gov/) by entering the code from the GenBank AAY34763.1 and the results of the search code were downloaded in FASTA format. After obtaining the data from NCBI databases, multiple sequence alignment was performed in order to find the representative envelope protein sequences to represent the entire population of DENV envelope protein data based on the similarity among sequences. This operation was executed using Clustal W2 software online via the website http://www.ebi.ac.uk/Tools/services /web_clustalw2/toolform.ebi. Out of the alignments, the 1OKE as GenBank code of the envelope protein, was pointed to the same structure and the researchers decided to take it as a representative. The structure gives Protein Model Portal code.
Protein model portal and protein data bank: PMP (Protein Model Portal) provides access to a variety of protein models that are computed by comparative modeling methods from different partner websites and it provide access to interactive services models and quality assessment as well. PMP was used to provide information about the sequence similarities. Protein models portal can be accessed via the website (www.proteinmodelportal.org/query/uniport/Q2XRR3). PMP provides a structure that is similar to the GenBank protein envelope of AAY34763.1 with 1OKE and 1OK8 as the PDB code structures. By entering the code 1OK8 PDB, the results showed similarities with other structures, namely 1OAN and 1OAM. The FASTA files of the three DENV envelope protein structure was taken to be re-aligned back by multiple sequence alignment with accessing the site http://www.ebi.ac.uk/Tools/services/web_clustalw2/toolform.ebi. The results from the alignments indicated that the sequence similarity of the envelope protein PDB code 1OKE have more than 50% similarity with 1OAM, 1OK8 and 1OAN. Determination of the structure using multiple sequence alignment was done afterter math. PDB code for a protein of the inhibition of ligand is 1OAN, because it has the same shape and cavity with 1OKE. While 1OAM was lastly used in the 1970s, according to the graph (http://www.rcsb.org/pdb) and 1OK8 only have 1 cavity.
Cavity on DENV envelope protein: Identifying binding sites or residues that form a cavity in the target structure is important in structure-based drug design (Kalyaanamoorthy and Chen, 2011). Research of (Yennamalli et al., 2009) shows the existence of a gap (cavity) that can be occupied by the ligand and affect the conformation of the DENV envelope protein. Cavity 33 is formed by these following amino acid residues: residues 1-8, 28, 30, 44, 151-155 and 316 of the peptide chain A, residues 97-109 and 244-247 of the peptide chain B.
Geometry optimization and energy minimization of the 3D structure of DENV envelope protein: The parameters were using default settings of MOE2008.10 (Tambunan et al., 2012). Minimization process aims for a conformational change in the structure of the envelope protein of DENV. Energy minimization was performed until the RMS gradient of 0.05 kcal / Å corresponding to protein (Vilar et al., 2008) .
Search of cyclical peptide ligands and the design of their 3D structure: The search of commercial cyclic peptide ligands was done online by accessing the website of the American Peptide Company http://www.americanpeptide.com/commerce/products_services/catalog/products.jsp, peptide company Mimotopes through http://www.mimotopes.com/ and the Bachem peptides company which can also be accessed online through the website http://shop.bachem.com/ep6sf/search.ep?gm=1andkeyWords=cyclo+peptide&categoryId=4750andsubmit=Search. The search towards the three companies found 301 commercial cyclic peptides that can be used for inhibiting the envelope protein. The three companies also provide information about the amino acid sequence, molecular formula and molecular weight.
Depiction of 3D structure cyclic peptide ligands: The depiction of a cyclic peptide ligands was done after the search of commercial cyclic peptide ligands of American Peptide, Mimotopes and bachem which gives the molecular formula ligand that can be performed using ACD / ChemSketch which then modeled in the form of 3D structures. The model peptide was stored with MDL molfile storage format. MDLmol file must be converted using the offline software MDLmol VegaZZ to be accessible in the MOE2008.10.
Geometry optimization and energy minimization of the ligand 3D structures: For the partial charge, the parameters used are AMBER99 method. It can be used for the validation of the position of the hydrogen atom in the ligand. Energy minimization process was done with RMS gradient 0.001 kcal / Å. Furthermore, now, the peptide ligands have been prepared for the next process, the molecular docking and molecular dynamics.
Molecular docking: The docking process of 301 commercial cyclic peptides as ligands and protein envelope was performed using MOE 2008.10. Based on the literature, to perform docking, the protein is rigid while the ligand is allowed to rotate (the so-called flexible docking). This is done so that the docking process goes like lock and key mechanism (Tambunan et al., 2011b; Ortigoza et al., 2012). Total pose used in the method of placement is 2,500,000.
Stages of refinement was performed for correcting the conformation of the ligand. Refinement forcefield is appropriate to the default configuration. Forcefield was chosen because it has a more accurate calculation. This is because the forcefield was using Generalized Born Solvation Model (GB/VI) at the stage of final energy evaluation gridmin while using electrostatic calculations on the minimization (Vilar et al., 2008; Moesa et al., 2012). The default setting of refinement force field was using a pocket cut off 6?, the distance receptors are being included in the process of docking. Retain is the final step in the process of docking. Retain set shows only one conformation of the most appropriate and the best of each ligand (Tambunan et al., 2011a).
Analysis of docking results: The next stage is the screening performed on 301 commercial cyclic peptides. Screening was done to see the best ligand of 301 ligands and compared with standard ligands. Ligand standard used is NITD448 Poh (Poh et al., 2009), KAMPMANN A5, A4 KAMPMANN (Kampmann et al., 2009), R1 Yennamali (Yennamalli et al., 2009) and C6 Wang (Wang et al., 2009). Screening was conducted in duplicate so that the first screening ligands obtained 50 best and the second screening also obtained 50 best ligands. The second screening of the data was also obtained with the same rank as much as 20 ligands and the remaining 60 ligands with different rank. Therefore, a third screening conducted at 80 ligand screening results from the first and second screening. The third screening resulted in 10 best ligands out of 80. Then, the best of 10 ligands was screened to the fourth to get the three best ligands by observing at MOE2008.10 ligand interactions. Of the three best ligands, ADME-Tox analysis was performed to obtain the best ligand.
Free energy association (ΔG): From the results of the 301 ligand screening with a standard for comparison, 10 best ligands were obtained as a fusion inhibitor candidates DENV envelope protein. The energy data can be seen in Table 1.
Table 1 shows that the 10 best ligand has ΔG0 values lower or negative than the standard so that the affinity of the top 10 commercial cyclic peptide ligands with DENV envelope protein are better when compared with standard ligands. In software MOE2008.10, Ki value is displayed as the value pKi with micromolar scale. The value of pKi can be used to determine the level of the formation stability of protein complexes and ligands. Indirectly, the value of pKi also related to the number of hydrogen bonds formed with residues.
| Table 1: | The 10 best Ligands ΔG°dan pKi Values |
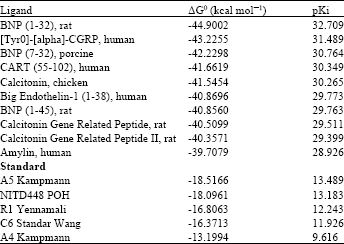 | |
Thus, more and stronger hydrogen bonds formed between the ligand with the protein, then value of the pKi will increase.
Commercial cyclic peptide ligand interactions with envelope protein: In addition to observing the value (ΔGbinding), interactions between ligands to proteins was also observed. There are other non-covalent interactions that can increase the affinity of the protein inhibitor. Hydrogen bonds are bonds that have a greater influence on the bond of the complex than others (Nurbaiti et al., 2010).
Table 2 shows that there are two out of 10 best ligands that can not be analyzed due to the big ligand size. The eight other ligands has more envelope protein interaction with the cavity than the standard ones. However, there are only three ligands can enter the cavity envelope protein as a whole. They are BNP (7-32) porcine, Big Endhotelin (1-38) human and Amylin human. Visualization of the eight ligands and the standards is described using MOE2008.10 in Fig. 1.
ADME-Tox prediction results of the commercial cyclic peptide ligands with envelope protein: The utilized online software is ACD-Labs and Toxtree-v2.5.0. In ACD-Labs, the ligands could be observed on their effects upon health of the organs, such as the liver, blood, gastrointestinal, cardiovascular, kidney and lungs.
| Table 2: | Top 10 ligand interactions based on ΔG0 with envelope proteins of molecular docking results |
 | |
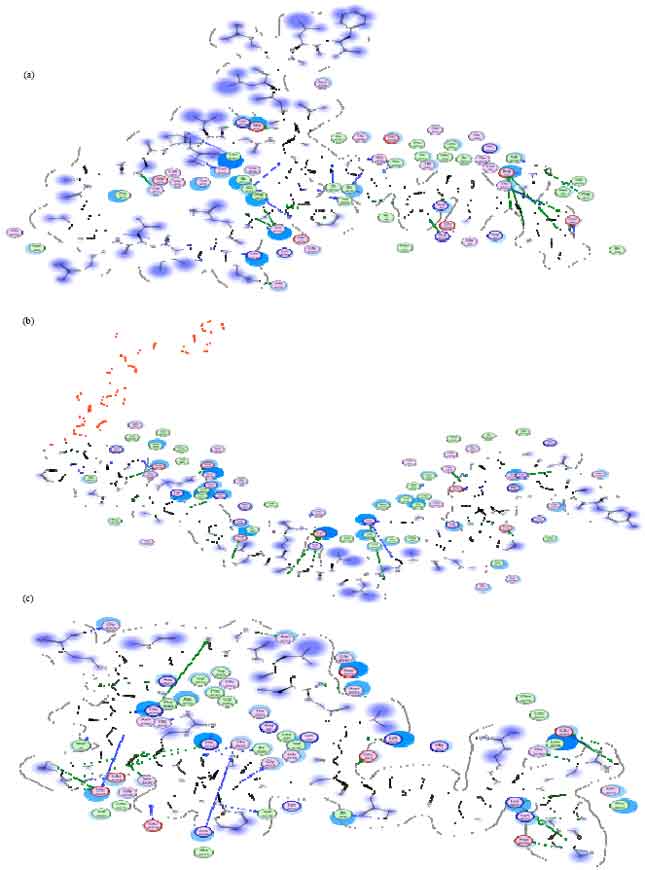  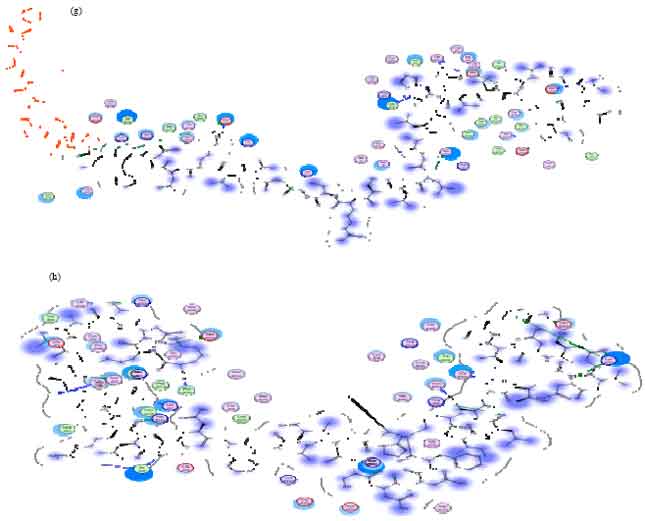 | |
| Fig. 1(a-h): | (a) Visualization of BNP (1-32), rat ligand interaction with protein envelope, (b) [Tyr0]-[Alpha]-CGRP, human ligand with protein envelope, (c) BNP (7-32), porcine ligand with protein envelope, (d) Calcitonin, chicken ligand with protein envelope, (e) Big endothelin-1 (1-38), Human ligand with protein envelope, (f) Calcitonin Gene Related Peptide, Rat Ligand with Protein Envelope, (g) Calcitonin gene related peptide II, rat ligand with protein envelope, (h) Amylin, human ligand with protein envelope |
Additionally, the bioavailability, active transport, toxicity were also observed in ACD-Labs. Ligands can also be observed on their negativity toward genotoxic or nongenotoxic carcinogenicity by using Toxtree-v2.5.0. This software can also be used to observe the potential of S. typhimurium TA100 mutagen and carcinogen based on Quantitative Structure Activity Relationship (QSAR). In ADME-Tox prediction of the ACD-Labs, it can be seen that the best ligands have little value oral bioavailability of less than 30%. Oral bioavailibility is the extent to which a drug or other materials will be available to the target tissue after administration of the drug or substance. Moreover, oligopeptides transporter 1 (PepT1) and apical sodium-dependent bile acid transporter (ASBT) is part of active transport in intestinal absorption. The results of active transport prediction of all commercial cyclic peptide ligands showed negative results and do not hinder the performance of PepT1 and ASBT so it can be excreted from the body. Ligand also can not penetrate the blood-brain barrier and does not cause interference with the central nervous system (Moroy et al., 2012; Bahadduri et al., 2010; Kortagere et al., 2008). On the probability of side effects, they show that the three best ligands have a large probability value of side effects and very potent to harm the blood, kidneys and liver. While in the cardiovascular, the best three ligands does not have significant probability of side effects. The ligands that has the least significant probability of side effects are BNP (7-32), porcine.
| Table 3: | The results of the ligand prediction through Toxtree-v2.5.0 |
 | |
The three best ligands, namely BNP (7-32) porcine, Big endothelin-1 (1-38) human, human Amylin and standard ligand, were calculated for their properties of toxicity using the set-Toxtree software v2.5.0. Toxtree is open source software that is able to estimate the molecular toxicology by computational means. In determining the toxicity level of a compound, the Toxtree-rules was based on the presence of a potentially mutagenic and carcinogenic properties in the test compound. Toxicity testing through Toxtree-v2.5.0 can be seen in Table 3.
From the results of predictive toxicity through Toxtree-v2.5.0, it was seen that the three ligands are in a negative parameter for nongenotoxic carcinogenity, S. typhiurium TA 100 mutagenic potential based on QSAR and a potential carcinogen based on QSAR properties in accordance with the expected toxicity. Prediction with Toxtree-v2.5.0 was conducted to analyze the nature of toxicity based on their carcinogenicity and mutagenicity properties, because both these properties have a direct impact on human health. After the prediction of the three ligands using ADME-Tox and o -v2.5.10 Toxtree, it was concluded that BNP (7-32), porcine is the best ligand. Thus, it will be evaluated using the method of molecular dynamics simulations with the condition of the hydrated molecules in a solvent.
Molecular dynamics: There are 3 stages in the molecular dynamics simulations, the initialization, equilibration and production. The initial stages are crucial to determine the initial state of the system such as atomic coordinates, speed and potential energy systems. After that, the simulation is run until the system reaches equilibration as indicated by the decline in potential energy of the systems. Equilibration is the position of atom density distribution in all parts of the system while providing a relaxed condition on the restrained molecule when the system is heated (Nurbaiti et al., 2010). The new atom positions are determined as a result in the production stage where the system has reached equilibrium (Vilar et al., 2008). Stages production yield trajectory of a simulation. The trajectory coordinates was formed by taking a snapshot of the changes over time that shows the state of each atom in the simulation period (Sharma et al., 2009).
Initialization timing: Molecular dynamics simulations carried out at a temperature of 300 K, for 100 ps. After molecular dynamics simulations, the determination of the correlation plot between the total potential energy versus simulation system, could be done. Determination of initialization was based on the duration in which the total potential energy of the system began to enter the equilibrium. After 30 ps, the envelope protein complex and the best ligand conformation was able to be adjusted with the solvent.
Molecular dynamics simulation at 310 K: Molecular dynamics simulation at a temperature of 310 K continued after the results of the initial phase for the envelope protein complex and the best ligand. The main simulation run during 5000 ps, then performed during the cooling stage for 10 ps. The room temperature of 300 K is usually used to perform molecular dynamics simulations. The next process is the cooling stage that was performed after the main simulations in order to find the lowest energy conformation of the molecule. The results of position, velocity and acceleration are saved every 0.5 ps. Molecular dynamics simulations was performed on the nanosecond time scale. It was due to the relative movement of the rigid body motion, i.e including movement of α-helical strands and the protein domains (Nurbaiti et al., 2010) .
The observed ligand interactions are contact residues and hydrogen bonding. Analysis of the residue contacts was conducted to determine the types of amino acid residues in proteins that interact with ligands. That’s because of non-covalent interactions that occur between proteins with ligand. It can increase the activity of the ligand to the protein. Residues cavity of the envelope protein interaction with the ligand are indicated by red text. During the simulation 5000 ps, BNP (7-32), porcine ligand consistently interacts with the fusion peptide segments and it can be seen that the ligand BNP (7-32), porcine also still interacts with residues cavity of the envelope protein in the cooling stage, i.e. with residues Glu 44, Gly 104, Asp 154, Val 151, Lys 246 and Lys 247. The residues of Ligand BNP (7-32), porcine that actively interact with the envelope protein are Glu 44, Gly 104, Asn 103, Lys 246 and Lys 247, at the time of docking simulations and dynamics simulation.
Molecular dynamics simulation at 312 K: Molecular dynamics simulations at 312 K temperature also showed changes in protein-ligand interactions. It shows that the residues from cavity of envelope protein, namely Val 151, Asp 154 and Lys 246, interact with the ligand BNP (7-32), porcine from initial stage to stage cooling.
Comparison of the interaction between the ligand BNP (7-32), porcine with the cavity of DENV envelope protein in a state of rigid molecular docking in a protein-ligand interactions in molecular dynamics simulations at a temperature of 312 K shows that the influence of solvation on the dynamic movement of the molecules will affect the affinity and stability of the bond between the ligand with the target protein.
Analysis of molecular dynamics simulations: The next step is to look at RMSD curves for observing the conformational changes of envelope protein and ligand BNP (7-32), porcine. It is shown at Fig. 2. RMSD values are useful to describe the magnitude of the conformational changes between the two coordinates of the atoms. It shows that the RMSD plot of simulation time that states the conformational changes between the envelope protein and ligand complexes BNP (7-32), porcine, during the molecular dynamics simulations at the moment.
It shows that protein-ligand interaction at a temperature of 310 and 312 K do not undergo significant changes. If the temperature rises, the interactions between proteins and ligands will be amended. Protein-ligand interactions will change if the structure of the protein changed as well. Complex BNP (7-32), porcine did not experience much conformational change. RMSD values at 310 and 312 K simulations also did not much differ, because there is a common protein structural changes in both temperature. However, ligand BNP (7-32), porcine is more interactive at 312 K than the temperature of 310 K.
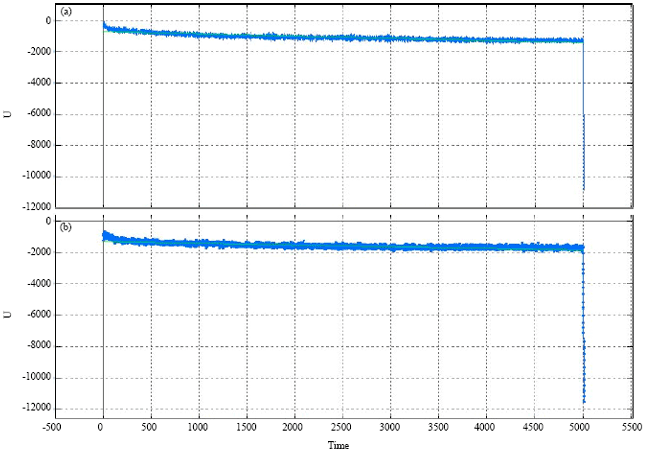 | |
| Fig. 2(a-b): | Production Curve during molecular dynamics simulation at 310K (a) Correlation plot d:/ penelitian/kinanty/dynamics/danamics 2-prod.mclb R: -0.5769 R2: 0.3329 (U): -0.138015 time-4722.59 and (b) Correlation plot d:/ penelitian/kinanty/dynamics/danamics 2-prod.mclb R: 0.4594 R2: 0.2111 (U): -0.10957 time-1302.39 312 K |
The results of the molecular dynamics simulation visualization for 5000 ps at the temperatures 310 K and 312 K shows that there are similarities of ligand interactions BNP (7-32), porcine with the binding on the envelope protein. It can also be seen with the curve overlapping at 5000 ps. During 2500-4500 ps, the RMSD value was changing at temperatures 310 and 312 K, indicating larger conformational structures-changes which occurred at 312 K more than 310 K. Overall it can be said that there is no significant difference in the structure of the conformational changes during molecular dynamics simulations at temperatures 310 and 312 K.
DISCUSSION
The in silico research that previously only deals with non-commercial agents, are now indeed immersed with the commercial compounds. Previously, our works show that the computational design for Dengue Drug was indeed feasible (Tambunan and Alamudi, 2010; Tambunan et al., 2009; Tambunan et al., 2011a, b). However, as the development of molecular virology are progressing forward, the existing target for dengue inhibitor must be updated, with our current research in the envelope protein of the Dengue virus itself.
The cyclic peptides have been proven as a stable lead compound for drugs (Bogdanowich-Knipp et al., 1999; Gudmundsson et al., 1999). In this end, it is indeed interesting for focusing on the development of cyclic peptides for Dengue virus inhibitors (Tambunan et al., 2011b). The potential of the cyclic peptide stability for drugs is our driving force for developing this in silico pipeline.
The recent availability of the commercial cyclic peptide databases are crucial for the completion of this research. The databases would eventually assist us in finding the right compounds in the screening process. Both Molecular Docking and Dynamics pipelines from MOE software have successfully providing us with the lead compound as drug candidate. The ADMET in silico determination has made this screening process much more rigid.
CONCLUSION
The commercial cyclic peptides were derived from the company's online site peptide. Then, the cyclic peptides were filtered to obtain the best ligands as inhibitors of envelope protein. The screening was conducted through molecular docking processes that produced 10 of the best ligands. They have ΔGbinding values lower than standard ligands. Moreover, the extensive screening were done among the best 10 ligand interaction analysis with envelope proteins through MOE2008.10 and produced three of the best ligands that can enter the cavity of the envelope protein. ADME-Tox analysis was done to observe the value of the toxicity of the three best ligands and obtained the best ligand, namely BNP (7-32), porcine. In the molecular dynamic simulation, during temperatures 310 and 312 K, ligand BNP (7-32), porcine can maintain interaction with the active site residues of the protein envelope until the end of the simulation at 5000 ps. From the analysis of the conformational changes at 310 and 312 K, it appears that the ligand BNP (7-32), porcine can maintain the stability of the protein-ligand complex conformations. Ligand BNP (7-32), porcine is more reactive at temperatures of 312 K compared to 310 K.
It is suggested to perform molecular dynamic simulations at temperatures 309K to study ligand-protein interactions in the healthy human body and to consider the possibility of a mutation in the protein due to the binding of the ligand. The wet lab experiments are necessary to further examine the influence of commercial cyclic peptide ligand inhibition against DENV infection levels both in vitro and in vivo.
ACKNOWLEDGMENT
The authors are grateful to Hibah Riset Unggulan DRPM UI No. DRPM/RII/224/RU-UI/2013 for funding this research. We are deeply indebted to Arli Aditya Parikesit and Usman Sumo Friend Tambunan for supervising this research and preparing the manuscript. We also extend our heartfelt gratitude to Kinanty for working on the technical details.
REFERENCES
- Adcock, S.A. and J.A. McCammon, 2006. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev., 106: 1589-1615.
CrossRefDirect Link - Bahadduri, P.M., J.E. Polli, P.W. Swaan and S. Ekins, 2010. Targeting drug transporters-combining in silico and In vitro approaches to predict In vivo. Methods Mol. Biol., 637: 65-103.
CrossRefDirect Link - Blondelle, S.E. and K. Lohner, 2010. Optimization and high-throughput screening of antimicrobial peptides. Curr. Pharm. Design, 16: 3204-3211.
CrossRef - Bogdanowich-Knipp, S.J., S. Chakrabarti, T.D. Williams, R.K. Dillman and T.J. Siahaan, 1999. Solution stability of linear vs. cyclic RGD peptides. J. Pept. Res., 53: 530-541.
Direct Link - Chen, A.A. and R.V. Pappu, 2007. Parameters of monovalent ions in the AMBER-99 forcefield: Assessment of inaccuracies and proposed improvements. J. Phys. Chem. B, 111: 11884-11887.
CrossRefDirect Link - Dubey, K.D., A.K. Chaubey and R.P. Ojha, 2011. Role of polarization in ligand docking and binding affinity prediction for inhibitors of dengue virus. Med. Chem. Res., 21: 1030-1038.
CrossRefDirect Link - Farias, K.J.S., P.R.L. Machado and B.A.L. Da Fonseca, 2013. Chloroquine inhibits dengue virus type 2 replication In vero cells but not in c6/36 cells. Sci. World J.
CrossRef - Gudmundsson, O.S., K. Nimkar, S. Gangwar, T.J. Siahaan and R.T. Borchardt, 1999. Phenylpropionic acid-based cyclic products of opioid peptides that exhibit metabolic stability to peptidases and excellent cellular permeation. Pharm. Res., 16: 16-23.
CrossRefDirect Link - Homeyer, N. and H. Gohlke, 2013. FEW-A workflow tool for free energy calculations of ligand binding. J. Comput. Chem.
CrossRef - Kalyaanamoorthy, S. and Y.P.P. Chen, 2011. Structure-based drug design to augment hit discovery. Drug Discovery Today, 16: 831-839.
CrossRefDirect Link - Kampmann, T., R. Yennamalli, P. Campbell, M.J. Stoermer, D.P. Fairlie, B. Kobe and P.R. Young, 2009. In silico screening of small molecule libraries using the dengue virus envelope E protein has identified compounds with antiviral activity against multiple flaviviruses. Antiviral Res., 84: 234-241.
CrossRefDirect Link - Karplus, M. and J.A. McCammon, 2002. Molecular dynamics simulations of biomolecules. Nature Structural Biol., 35: 646-652.
CrossRefDirect Link - Kielian, M., C. Chanel-Vos and M. Liao, 2010. Alphavirus entry and membrane fusion. Viruses, 2: 796-825.
Direct Link - Kortagere, S., D. Chekmarev, W.J. Welsh and S. Ekins, 2008. New predictive models for blood-brain barrier permeability of drug-like molecules. Pharm. Res., 25: 1836-1845.
CrossRefDirect Link - Lee, Y.K., S.K. Tan, H.A. Wahab, R. Yusof and N.A. Rahman, 2007. Nonsubstrate based inhibitors of dengue virus serine protease: A molecular docking approach to study binding interactions between protease and inhibitors. Asia Pacific J. Mol. Biol. Biotechnol., 15: 53-59.
Direct Link - Majeux, N., M. Scarsi and A. Caflisch, 2001. Efficient electrostatic solvation model for protein-fragment docking. Proteins, 42: 256-268.
CrossRefDirect Link - Moesa, H.A., S. Wakabayashi, K. Nakai and Ashwini, 2012. Chemical composition is maintained in poorly conserved intrinsically disordered regions and suggests a means for their classification. Mol. Bio. Syst., 8: 3262-3273.
CrossRefDirect Link - Moroy, G., V.Y. Martiny, P. Vayer, B.O. Villoutreix and M.A. Miteva, 2012. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discovery Today, 17: 44-55.
CrossRefDirect Link - Noble, C.G., Y.L. Chen, H. Dong, F. Gu and S.P. Lim et al., 2010. Strategies for development of dengue virus inhibitors. Antiviral Res., 85: 450-462.
CrossRefDirect Link - Nurbaiti, S., H. Nagao, H. Saito, R. Hertadi, M.A. Martoprawiro and Akhmaloka, 2010. Domain motions of Klenow-like DNA polymerase I ITB-1 in the absence of substrate. Int. J. Integr. Biol., 9: 104-110.
Direct Link - Ortigoza, M.B., O. Dibben, J. Maamary, L. Martinez-Gil and V.H. Leyva-Grado et al., 2012. A novel small molecule inhibitor of influenza A viruses that targets polymerase function and indirectly induces interferon. PLoS Pathog., Vol. 8.
CrossRef - Poh, M.K., A. Yipa, S. Zhanga, J.P. Priestlee and N. Ling Ma et al., 2009. A small molecule fusion inhibitor of dengue virus. Antiviral Res., 84: 260-266.
CrossRefDirect Link - Sampath, A. and R. Padmanabhan, 2009. Molecular targets for flavivirus drug discovery. Antiviral Res., 81: 6-15.
CrossRefDirect Link - Samsa, M.M., J.A. Mondotte, N.G. Iglesias, I. Assuncao-Miranda and G. Barbosa-Lima et al., 2009. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog., Vol. 5.
CrossRef - Sharma, R.D., A.M. Lynn, P.K. Sharma, Rajnee and S. Jawaid, 2009. High temperature unfolding of Bacillus anthracis amidase-03 by molecular dynamics simulations. Bioinformation, 3: 430-434.
Direct Link - Shu, M., Z. Lin, Y. Zhang, Y. Wu, H. Mei and Y. Jiang, 2011. Molecular dynamics simulation of oseltamivir resistance in neuraminidase of avian influenza H5N1 virus. J. Mol. Modeling, 17: 587-592.
CrossRefDirect Link - Tambunan, U.S.F., N. Apriyanti, A.A. Parikesit, W. Chua and K. Wuryani, 2011. Computational design of disulfide cyclic peptide as potential inhibitor of complex NS2B-NS3 dengue virus protease. Afr. J. Biotechnol., 10: 12281-12290.
Direct Link - Tambunan, U.S.F., Fadilah and A.A. Parikesit, 2010. Bioactive compounds screening from zingiberaceae family as influenza a/swine flu virus neuraminidase inhibitor through docking approach. Online J. Biol. Sci., 10: 151-156.
Direct Link - Tambunan, U.S.F., R. Harganingtyas and A.A. Parikesit, 2012. In silico modification of (1R, 2R, 3R, 5S)-(-)- isopinocampheylamine as inhibitors of m2 proton channel in Influenza A virus subtype H1N1, using the molecular docking approach. Trends Bioinfo., 5: 25-46.
CrossRefDirect Link - Tambunan, U.S.F., R.S. Noors, A.A. Parikesit, Elyana and W. Ronggo, 2011. Molecular dynamics simulation of DENV RNA-dependent RNA-polymerase with potential inhibitor of disulfide cyclic peptide. J. Biol. Sci., 11: 48-62.
Direct Link - Tambunan, U.S.F., A.A. Parikesit, Hendra, R.I. Taufik and F. Amelia, 2009. In silico analysis of envelope dengue virus-2 and envelope dengue virus-3 protein as the backbone of dengue virus tetravalent vaccine by using homology modeling method. Online J. Biol. Sci., 9: 6-16.
Direct Link - Tambunan, U.S.F. and S. Alamudi, 2010. Designing cyclic peptide inhibitor of dengue virus NS3-NS2B protease by using molecular docking approach. Bioinformation, 5: 250-254.
Direct Link - Teissier, E., F. Penin and E.I. Pecheur, 2011. Targeting cell entry of enveloped viruses as an antiviral strategy. Molecules, 16: 221-250.
CrossRefDirect Link - Tomlinson, S.M., R.D. Malmstrom and S.J. Watowich, 2009. New approaches to structure-based discovery of dengue protease inhibitors. Infect. Disorders Drug Targets, 9: 327-343.
Direct Link - Vilar, S., G. Cozza and S. Moro, 2008. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem., 8: 1555-1572.
CrossRefDirect Link - Wang, Q.Y., S.J. Patel, E. Vangrevelinghe, H.Y. Xu and R. Rao et al., 2009. A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother., 53: 1823-1831.
CrossRefDirect Link - Yennamalli, R., N. Subbarao, T. Kampmann, R.P. McGeary, P.R. Young and B. Kobe, 2009. Identification of novel target sites and an inhibitor of the dengue virus E protein. J. Comput. Aided Mol. Design, 23: 333-341.
CrossRefDirect Link