
Amad M. Al-Azzawi
Ras Al Khaimah College of Pharmaceutical Sciences, Ras Al Khaimah Medical and Health Sciences University, Ras Al Khaimah, P.O. Box 11172, UAE
Pakistan Journal of Biological Sciences
Year: 2012 | Volume: 15 | Issue: 2 | Page No.: 92-97







ABSTRACT
Tecoma stans (Bignoniaceas) is a central and south American tree used for the control of diabetes. This plant is cultivated in Iraq. The dried leaves were soaked in ethanol and water separately for 3 days then filtered and dried. The genotoxic potential of Tecoma stans was studied by in vivo and in vitro system. This study examined the genotoxic activity of aqueous and ethanolic extracts on bone marrow cells from BALB/c mice through evaluation of mitotic index and chromosomal aberrations and cytotoxic effect of the two extracts on Mouse Embryo Fibroblast (MEF) cell line. No alteration in the total number of chromosomal aberrations or the number of cells with chromosomal aberrations observed and percentage of mitotic index at the concentrations tested remained unchanged. The higher concentrations used of the plant extracts had a cytotoxic effect on the MEF cell line. Both extracts had no significant clastogenic effect in vivo but showed cytotoxic effects on mouse embryo in vitro, caution should be exercised in the use of this substance as a medicine.
PDF Abstract XML References Citation
Received: September 30, 2011;
Accepted: December 15, 2011;
Published: February 16, 2012
How to cite this article
Amad M. Al-Azzawi, 2012. Genotoxic and Cytotoxic Study of Tecoma stans Bignoniaceae. Pakistan Journal of Biological Sciences, 15: 92-97.
DOI: 10.3923/pjbs.2012.92.97
URL: https://scialert.net/abstract/?doi=pjbs.2012.92.97
DOI: 10.3923/pjbs.2012.92.97
URL: https://scialert.net/abstract/?doi=pjbs.2012.92.97
INTRODUCTION
Tecoma stans, a common in Latin America, is a plant traditionally used in Mexico for the control of diabetes. The presence of alkaloids in Tecoma stans was first reported in 1899 (Jones et al., 1963). Isolated alkaloids and evaluated there antidiabetic in vivo and in vitro activity (Costantino et al., 2003). Marzouk et al. (2006) identified a number of compounds from the fruits and flowers of the tecoma which had antioxidant activity and anti-proliferative effect against cancer cell lines. In 2009 a study focuses on elucidating the pharmacological mechanism of Tecoma stans antidiabetic effect of the aqueous extract on type 2 diabetes mellitus (DM2) animal models is due to intestinal alpha-glucosidase inhibition by decreasing the postprandial hyper-glycaemia peak; in addition, Tecoma stans aqueous extract sub-chronic administration reduces triglycerides and cholesterol, without modifying fasting glucose (Aguilar-Santamaria et al., 2009). Recently Alonso-Castro et al. (2010) found that the aqueous extract Tecoma stans exert their antidiabetic effects stimulating glucose uptake in both insulin-sensitive and insulin-resistant murine and human adipocytes without significant proadipogenic or antiadipogenic side effects. Since Iraq is using traditional medicine throughout history and modern life for the treatment of a variety of diseases, consequently the plant is newly cultivated in Iraq and investigated for the first time. The aqueous extract of plant showed potential anti-diabetic activity and systematic literature survey revealed that studies on the genotoxicity of Tecoma stans are not available. The genotoxic potential of Tecoma stans was studied through in vivo and in vitro system.
MATERIALS AND METHODS
Collection of plant material: The leaves of Tecoma stans Juss (Bignoniacease) cultivated in Iraq were collected from the area of University of Baghdad (Al-Jadria) in August and November 2003. The specimen was authenticated by Dr. Ali Al-Mousawi (University of Baghdad college of Science department of Biology) the leaves were dried at room temperature in the shade, pulverized by mechanical mills and weighed.
Distilled water and ethanol extraction: Fifty grams of the dried leaves were pulverized then soaked with each solvent separately for 72 h at 25°C with occasional stirring (protected from light). The extract was filtered and then the solvent was removed under reduced pressure to dryness by rotary evaporator. The recovery weight was about 20-30% from the dried material (Tan et al., 2005; Costa-Lotufo et al., 2005).
Animals: Eight weeks old male BALB/c mice weighing (22±2 g) from the University of Baghdad, College of Science, Department of Biology. The mice were kept in metallic cages at a mean temperature of 23°C at a 12 h dark-light cycle and permitted to freely consume water and food in the animal house of Research Center of Biotechnology Al-Nahrain University. All experiments were conducted between 10.00 and 17.00 h and were in accordance with the ethical guidelines of the International association for study of pain (Zimmerman, 1983) and approved by the institutional animal ethics committee.
Experimental design: The experimental design was performed by randomly dividing the 60 male mice into two main groups for each extract and then 6 equal experimental groups (five mice per group) were used for these determinations. Ethanolic and aqueous extracts of Tecoma stans previously prepared were used at concentrations of 0.25, 2.5, 25 and 250 mg mL-1 for each group respectively. Each group received the plant extract via oral gavage suspended in phosphate buffer solution PBS for fifteen consecutive days. While the control group received PBS only in a fixed volume of 1 mL 100 g-1 body weight the sixth group received doxorubicin (DX) 10 mg kg-1 as positive control (sigma). Three independent experiments were performed (Paniagua et al., 2005).
Bone marrow chromosome assay: The animal was injected with 0.25 mL of colchicine (Al-Hikma Pharmaceuticals, Jordan) with a concentration of 0.6 mg mL-1 intraperitoneally (IP) 2 h before killing the animal. Then animal was sacrificed by cervical dislocation under mild chloroform anesthesia. The animal was fixed on vernal side against the anatomy plate and the abdominal side of the animal and its thigh region was swabbed with 70% ethanol. The thigh bone was then taken and cleaned from the other tissues and muscles, then gapped from the middle with a forceps in a vertical position over the edge of the test tube by a sterile syringe. Five mililiter of PBS (Phosphate Saline Buffer) was injected to wash and drop the bone marrow in the test tube.
The test tube was taken and put in the centrifuge at speed of 2000 rpm for 10 min. The supernatant was removed and 5 mL of KCl (0.075 M) was pre-warmed at 37°C and added as a hypotonic solution. The test tubes were left for 30 min in the water bath at 37°C with occasional shaking. The samples were centrifuged at 2000 rpm for 10 min. Later, the supernatant was removed and the fixative solution (methanol: glacial acetic acid 3:1) was added drop wise on the inside wall of the test tube with the continuous shaking; the volume was fixed to 5 mL.
The tubes were kept at 4°C for 30 min to fix the cells. The tubes were then centrifuged at 2000 rpm for 10 min. The process was repeated for 3 times and the cells were suspended in 2 mL of the fixative solution.
By a Pasture pipette few drops from the tube were dropped vertically on the slide from a height of 60 cm at a rate of 4-5 drops to give the chance for the chromosomes to spread well. Later, the slides were dried on a hot plate at 50°C for 1 min. The slides were stained with Giemsa stain (The stain was prepared by mixing the following chemicals in 40 mL of D.W.; Original Giemsa Stain stock 1 mL, Methanol 1.25 mL and Sodium bicarbonate solution 1 mL) and left for 4 min and then washed with D.W.
Metaphase cell preparations were obtained from bone marrow cells by the technique of Burim et al. (1999) and Ford and Hamerton (1956) with some modifications. One hundred metaphases per animal were analyzed to determine the frequency of chromosomal aberrations. The mitotic index represented the number of metaphase cells detected in 2000 cells analyzed per animal and was expressed as a percentage. Three slides for each animal were prepared for cytogenetic assay.
Mitotic index assay (MI): The slides were examined under high power (40 magnification power) of compound light microscope and 1000 of divided and non- divided cells were counted and the percentage rate was calculated for only the divided ones according to the following equation:
where, the total number of cells equals 1000 (Mohammed et al., 2009; Shubber and Juma, 2004).
Chromosomal aberration assay (CA): The prepared slides were examined under the oil immersion lens under high power (100 magnification power) for 100 divided cells per each animal and the cells should be at the metaphase stage of the mitotic division where the chromosomal aberrations are clear (Maranon et al., 2004; Cunha et al., 1997).
Mouse Embryo Fibroblast (MEF): Cell line was established and kindly provided from Iraqi Center for Cancer and Medical Genetics Research (ICCMGR) (Al-Shamery, 2003). Cell lines were maintained as described by Freshney (Freshney, 1994).
Viable cell count: Cell counting was done by the use of Trypan blue dyes were the dead cell will take the dye: (0.2 mL of the dye with 0.2 mL of the cell with 1.6 mL of PBS).Then the counting was done by using improved double Neubauner ruling chamber (Freshney, 1994).
Cytotoxic assay of extract on cancer cell lines: All extracts were dissolved 5% (DMSO) (Dimethyle sulf oxide BDH.England) sterilized with a milipour filter (0.22 μm) then the preparation of 8 different concentations as following:
a) 0.1, b) 0.05, c) 0.025, d) 0.0125, e) 0.00625, f) 0.003125, g) 0.0015625, h) 0.0007812 g = 0.7812 mg = 781.2 μg mL-1. All dilutions made to give 0.1% DMSO final concentration.
| • | The bottles with the culture were treated with trypsin /veresen solution then 20 mL of SFM was added to the culture bottles with the culture and mixed |
| • | Then 0.2 mL of the mixture was transported to 96- well microplates contain at each well 1x105 cell per well and were allowed to adhere for16 h before treatment |
| • | Then 0.2 mL of the aliquote solution of the tested compound were added to the cultured cells and the whole system was incubated for 72 h at 37°C and humidified 5% CO2 atmosphere |
| • | At the end of the incubation time, cells were washed with PBS Cell viability was measured after removing the medium adding 100 mL of 5 mg mL-1 solution of crystal violet (BDH-England) and incubated for 20 min at 37°C. Then after removing the crystal violet/ formalin solution cells were washed with PBS |
| • | The absorbency was determined using a micro plate reader (organon Teknika reader 2305, Australia) at 492 nm (test wave length) the assay was performed triplicate fashion (Freshney, 1994; Mahony et al., 1989) |
| • | The inhibiting rate of all growth (the percentage of cytotoxicity) was calculated as A-B/Ax100 Where A is the mean optical density of untreated wells and B is the optical density of the treated wells (Chiang et al., 2003; Mather and Roberts, 1998) |
| • | The antineoplastic agent etoposide, was taken as positive control |
Statistical analysis: Analysis of data was performed by using SPSS (version 15). Results expressed as mean±S.E. Statistical differences were determined by Student Newman Keul test for multiple comparison after ANOVA.
RESULTS AND DISCUSSION
Mitotic index and chromosomal aberration: The dose dependent effects of the aqueous and ethanolic plant extract on the division of bone marrow cells of the BALB/c mice showed that as the dose increases there will be no significant alteration in the mitotic index and the number of chromosomal aberration (Table 1 and 2).
The chromosomal aberrations seen in bone marrow cells from BALB/c mice treated with aqueous and ethanolic plant extract were chromatid gaps and chromatid break. Where the plant extract did not alter the total number of chromosomal aberrations or the number of cells with chromosomal aberrations, at the concentrations tested. The vehicle control group of mice showed a few aberrant metaphases and a few aberrations per hundred metaphases.
| Table 1: | The mitotic index and the number of chromosomal aberration in bone marrow cells from BALB/c mice treated by aqueous plant extract |
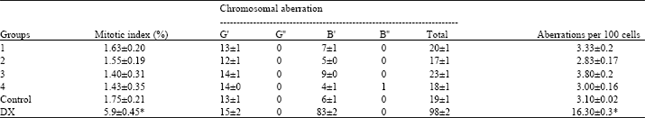 | |
| One hundred cells were analyzed per animal for a total of 600 cells per treatment. G’: Chromatid gap; G”: Chromosome gap; B’: Chromatid break; B”: Chromosome break; DX: Doxorubicin, *Statistically significant difference with respect to the result obtained in the negative control group. ANOVA and Student t tests, p≤0.05 | |
| Table 2: | The mitotic index and the number of chromosomal aberration in bone marrow cells from BALB/c mice treated by ethanolic plant extract |
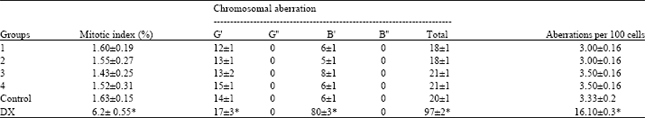 | |
| One hundred cells were analyzed per animal for a total of 600 cells per treatment. G’: Chromatid gap; G”: Chromosome gap; B’: Chromatid break; B”: Chromosome break; DX: Doxorubicin, *Statistically significant difference with respect to the result obtained in the negative control group. ANOVA and Student t tests, p≤0.05 | |
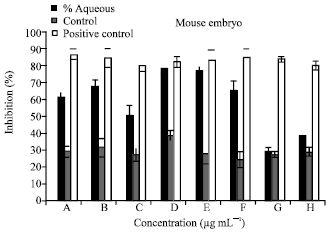 | |
| Fig. 1: | The percentage of inhibition of MEF cell line treated with aqueous plant extract with the following concentrations, 100000 (A), 50000 (B), 25000 (C), 12500 (D), 6250 (E), 3125(F), 1562 (G), 7812 (H) μg mL-1 |
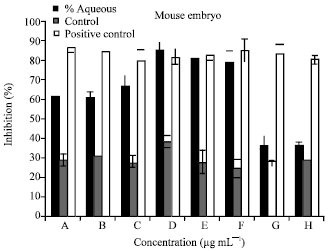 | |
| Fig. 2: | The percentage of inhibition of MEF cell line treated with alcoholic plant extract with the following concentrations, 100000 (A), 50000 (B), 25000 (C), 12500 (D), 6250 (E), 3125(F), 1562(G),7812 (H) μg mL-1 |
The positive control group showed a large number of aberrant metaphases and statistically significant number of chromosome aberrations when compared to the vehicle control group.
Cytotoxicity: The cytotoxicity effects of different concentrations of alcoholic and aqueous extracts 100000(A), 50000(B), 25000(C), 12500(D), 6250(E), 3125(F), 1562.5(G), 781.2(H) μg mL-1 were tested on normal cells line MEF were illustrated in (Fig. 1 and 2). The concentrations of the aqueous extracts 12500 and 6250 μg mL-1 achieved high percentage of inhibition of 78.64583 and 76.61871%, respectively. While cells treated with low concentrations of aqueous extracts 781.2 μg mL-1 showed low level of toxicity giving percentage of inhibition of 38.88889%.
As for the alcoholic extract the 12500 and 6250 μg mL-1 achieved high percentage of inhibition of 85.67708 and 81.65468%, respectively. While cells treated with low concentrations of alcoholic extracts 781.2 μg mL-1 showed low level of toxicity giving percentage of inhibition of 36.11111%.
Morphological assessment using inverted microscopy of the cultured cells indicated that cell death occurred in the cell lines. The death is shown in the form of increase in number of vacuoles with condensation of nuclear chromatin and, membrane changes were confirmed. In addition cell disintegration and reduction in cell number were seen. While the untreated controls, the cells appeared morphologically normal so the active constitutes in the plant extract causes morphological changes and cell death.
Genotoxic studies are useful to identify the level of DNA damage induced by plant extracts, as well as to give a clue about the possible clinical consequences of human exposure. In vivo acute studies in mammalian organisms, in particular, are usually considered the last type of assays in a hierarchical test battery designed to determine the genotoxic potential of an plant material; in this step, high doses of the tested compound are usually included; however, new studies are needed to analyze risk assessment that is, incorporating variables such as sex, age, time, dose or route of administration, to better understand the toxic potential as well as to have more data for a reasonable extrapolation to human exposure.
The presence of polyphenolic, flavonoid and β- Sitosterol compounds in the aqueous and alcoholic extracts of Tecoma stans, may explain the cytotoxic activity against (MEF) cell lines used. These compounds have been reported to display some relevant biological activities such as antibacterial, antiviral, immune-stimulating and estrogenic effects and antiproliferative and cytotoxic properties in several tumor cells (Hamburger and Hostettmann, 1991; Beltrame et al., 2002; Suffredimi et al., 2004).
The Tecoma stans is a plant with a potential antidiabetic activity that is currently being tested for their possible therapeutic use. Evaluation of the cytotoxic and mutagenic potential of plant extract on mouse embryo and in BALB/c mouse bone marrow cells is important because of the biological properties of this extract. In this study, the use of doses was based on the maximum amount of plant extracted which can be given practically to the mouse in each dose since no toxicity dose was established from literature till date.
Some amount of cell death found in untreated cells is due to natural cell death. This might be caused by nutrient depletion in growth media or contact inhibition.
The 250 mg mL-1 was the highest dose given to the mouse during the genotoxic test did not induce an increase in the mitotic index nor chromosomal aberrations of the mouse, indicating no in vivo genotoxic potential of the plant extracts. Where the frequency of chromosomal aberrations in the negative controls was 3.33-3.1/100 cells, which was within the range of 0-6.7% for spontaneous aberrations proposed by Kasuba et al. (1995). From our results we see the chromosomal aberrations for both extracts within the normal values. Concentrations of both plants extracts showed no effect on the MI.
The cytotoxic effect of both extracts at different concentration were significant because the first six concentrations used showed an inhibitory effect at or more than 50%. The cytotoxic effect of both extracts at concentration of 12500 μg mL-1 on the mouse embryo is five times less than both aqueous and alcoholic extracts used in the evaluation of mitotic index and chromosomal aberrations.
The MI of BALB/c mice showed that both extracts did not interfere with the growth and division of bone marrow cells. At the highest dose tested, no deaths were recorded. The negative results with both extracts in the assays in vivo may indicate that the extracts were bio-transformed in the liver, leading to their inactivation or that there was rapid excretion of the metabolite with no effect on the bone marrow cells. In this regard, little is known of the pharmacokinetics of plant extracts in mice. It is also possible that bone marrow cells may not be the target organ for this compound.
CONCLUSION
Both extracts had no significant clastogenic effect in vivo. However, since the compound did show cytotoxic effects on mouse embryo in vitro, caution should be exercised in the use of this substance as a medicine. However, future tests to measure genetic damage, as well as chronic exposure to the extracts, should be performed to have a better conclusion on the agent’s genotoxicity.
ACKNOWLEDGMENTS
I am grateful to Prof. Ekbal Al-Khateeb College of Pharmacy University of Baghdad, Assist. Prof. Kulood Al-Sameraei head of the Al- Nahrain biotechnology research center Al-Nahrain University for their guidance and engorgement, also I thank Dr.Mohammed Al-Jumaily Al-Nahrain biotechnology research center Al-Nahrain University and Dr. Al-Shamery, A.M.H in the Iraqi center for cancer and Medical genetic research, for valuable assistance.
REFERENCES
- Marzouk, M., A. Gamal-Eldeen, M. Mohamed and M. El-Sayed, 2006. Anti-proliferative and antioxidant constituents from Tecoma stans. Z. Naturforsch. C., 61: 783-791.
PubMedDirect Link - Alonso-Castro, A.J., R. Zapata-Bustos, J. Romo-Yanez, P. Camarillo-Ledesma, M. Gomez-Sanchez and L.A. Salazar-Olivo, 2010. The antidiabetic plants Tecoma stans (L.) Juss. ex Kunth (Bignoniaceae) and Teucrium cubense Jacq (Lamiaceae) induce the incorporation of glucose in insulin-sensitive and insulin-resistant murine and human adipocytes. J. Ethnopharmacol., 127: 1-6.
CrossRefPubMedDirect Link - Tan, M.L., S.F. Suleiman, N. Najimuddin, M.R. Samian and T.S. Tengku-Muhammad, 2005. methanolic extract of pereskia bleo (kunth) dc. (cactaceae) induces apoptosis in breast carcinoma, t47-d cell line. J. Ethnopharmacol., 96: 287-294.
Direct Link - Costa-Lotufo, L.V., M.T.H. Khan, A. Ather, D.V. Wilke and P.C. Jimenez et al., 2005. Studies of the anticancer potential of plants used in Bangladeshi folk medicine. J. Ethnopharmacol., 99: 21-30.
CrossRefPubMedDirect Link - Paniagua, P.R., M.E. Bujaidar, R.S. Cadena, M.D. Jasso and G.J. Perezet al., 2005. Genotoxic and cytotoxic studies of β-sitosterol and pteropodine in mouse. J. Biomed. Biotech., 2005: 242-247.
Direct Link - Ford, C.E. and J.L. Hamerton, 1956. A colchicine, hypotonic-citrate, squash sequence for mammalian chromosomes. Stain Technol., 31: 247-251.
PubMedDirect Link - Burim, R.V., R. Canalle, J.L.C. Lopes and C.S. Takahashi, 1999. Genotoxic action of the sesquiterpene lactone glaucolide B on mammalian cells in vitro and in vivo. Genet. Mol. Biol., 22: 401-406.
CrossRefDirect Link - Mohammed, B.M., K.J. Karim and N.Y. Yaseen, 2009. Anti mutagenic effects of thymus syriacus extract against the genotoxicity of gemcitabine in male albino mice. J. Duhok Univ., 12: 216-226.
Direct Link - Cunha, R.A.F., C.P. Koiffman, D.H. Souza and K. Takei, 1997. Clastogentic effects of different Ureaplasma urealyticum serovars on human chromosomes. Braz. J. Med. Biol. Res., 30: 749-757.
CrossRefDirect Link - Mahony, D.E., E. Gilliat, S. Dawson, E. Stockdale and S.H. Lee, 1989. Vero cell assay for rapid detection of clostridium perfringens enterotoxin. Applied Environ. Microbiol., 55: 2141-2143.
Direct Link - Chiang, L.C., W. Chang, M.Y. Chang and C.C. Lin, 2003. In vitro cytotoxic, antiviral and immunomodulatory effects of Plantago major and Plantago asiatica. Am. J. Chin. Med., 31: 225-234.
CrossRefPubMedDirect Link - Hamburger, M. and K. Hostettmann, 1991. Bioactivity in plants: The link between phytochemistry and medicine. Phytochemistry, 30: 3864-3874.
CrossRefDirect Link - Beltrame, F.L., G.L. Pessini, D.L. Doro, B.P.D. Filho, R.B. Bazotte and D.A.G. Cortez, 2002. Evaluation of the antidiabetic and antibacterial activity of Cissus sicyoides. Braz. Arch. Biol. Tech., 45: 21-25.
Direct Link - Suffredimi, I.B., H.S. Sader, A.G. Goncalves, A.O. Reis, A.C. Garles, A.D. Varella and R.N. Younes, 2004. Screening of antibacterial extracts from plants nature to the Brazilian amazon rain forest. Braz. J. Med. Biol. Res., 37: 379-384.
CrossRefDirect Link - Zimmermann, M., 1983. Ethical guidelines for investigations of experimental pain in conscious animals. Pain, 16: 109-110.
CrossRefPubMedDirect Link - Kasuba, V., K. Sentija, V. Garaj-Vrhovac and A. Fucic, 1995. Chromosome aberrations in peripheral blood lymphocytes from control individuals. Mutation Res. Lett., 346: 187-193.
CrossRefPubMedDirect Link