
T.S. Bonny
Department of Microbiology, University of Dhaka, Dhaka-1000, Bangladesh
N. Azmuda
Department of Microbiology, University of Dhaka, Dhaka-1000, Bangladesh
S.I. Khan
Department of Microbiology, University of Dhaka, Dhaka-1000, Bangladesh
N.K. Birkeland
Department of Biology, University of Bergen, P.O. Box 7803, N-5020, Bergen, Norway
M.Z. Rahman
Virology Laboratory, Laboratory Science Division, International Centre for Diarrhoeal Diseases Research, P.O. Box 128, Dhaka-1000, Bangladesh
Pakistan Journal of Biological Sciences
Year: 2010 | Volume: 13 | Issue: 19 | Page No.: 937-945







ABSTRACT
This research involved an environmental strain of Stenotrophomonas maltophilia which has been reported to produce serological cross-reactivity with Shigella dysenteriae type 8 specific antisera. Since clinical diagnosis of shigellosis is largely based on culture and serology, the investigation was aimed at in vivo and in vitro virulence comparison between the culturally similar environmental S. maltophilia isolate and the reference S. dysenteriae strains. The findings of this study revealed the absence of virulent genes of Shigella sp. like ipaH, virA and stx1 and characteristic invasive large plasmid in the test isolate. The Western blot analysis revealed that serological cross-reactivity of Stenotrophomonas maltophilia was due to certain protein component(s) in its outer membrane. The isolate was capable of producing extracellular protease, exhibited alpha hemolysis and was negative for hemagglutinating assay. The isolate gave negative reaction with rabbit ileal loop and Sereny tests. The S. maltophilia isolate did not possess any enterotoxic or invasive property as that of virulent S. dysenteriae strains. Further characterizations and adequate genetic manipulations of this environmental isolate may contribute to the development of a potential vaccine candidate for shigellosis.
PDF Abstract XML References Citation
How to cite this article
T.S. Bonny, N. Azmuda, S.I. Khan, N.K. Birkeland and M.Z. Rahman, 2010. Virulence of Environmental Stenotrophomonas maltophilia Serologically Cross-reacting with Shigella-specific Antisera. Pakistan Journal of Biological Sciences, 13: 937-945.
DOI: 10.3923/pjbs.2010.937.945
URL: https://scialert.net/abstract/?doi=pjbs.2010.937.945
DOI: 10.3923/pjbs.2010.937.945
URL: https://scialert.net/abstract/?doi=pjbs.2010.937.945
INTRODUCTION
Shigellosis or bacillary dysentery caused by Shigella sp. is endemic in many developing countries including Bangladesh. Among at least 80 million cases, 700,000 deaths occur each year due to shigellosis in developing countries. Seventy percent of these cases occur in children less than 5 years of age (WHO, 2005). By the age of 2, about 37% of the children in Bangladesh have acquired the infection (Seidlein et al., 2006). The poor sanitary conditions in developing countries like Bangladesh contribute to the spread of the disease and increasing antibiotic resistance further complicates the treatment. A safe and effective Shigella vaccine offers great potential as a means of preventing shigellosis.
Laboratory diagnosis of Shigella is often based on the isolation of the organism from feces of the patients by means of cultural and biochemical characteristics (Kelly et al., 1985). The slide agglutination test using a number of commercially available antisera specific to different groups and types, is a common and indispensable test for serological typing of Shigella sp. (Ewing and Lindberg, 1984; Evins et al., 1988).
Bacterial strains carrying identical or similar antigenic components on their surface structure might also react with the antibodies produced against other strains (Grubor et al., 2006; Houpikian and Raoult, 2003; Khan et al., 2003). It was found that various types of microorganisms such as Hafnia alvei, Plesiomonas shigelloides, Providencia alcalifaciens and Yersinia enterocolitica serotype 03 and E. coli (O114:H32, O157:H7, O157:H19, etc.) gave serological cross-reactivity with polyclonal group-specific Shigella antisera (Lefebvre et al., 1995). A number of environmental isolates were also found to cross-react with different types of Shigella-specific monovalent antisera. But the major cellular components giving rise to such cross-reactivity remains to be identified. Rahman et al. (2007) demonstrated that various environmental isolates cross-reacted with six different serotypes of Shigella and were, in each case, highly type-specific. Amongst those isolates, two environmental S. maltophilia exhibited significant serological cross-reactivity with S. dysenteriae type 8 specific monovalent antisera. These environmental isolates producing a high cross-reactivity with various types of Shigella antisera represent a potential for development of a shigellosis vaccine based on live cells, because they probably are not virulent like clinical organisms (Rahman et al., 2007).
The present investigation involved the comparison of virulent properties of the previously reported environmental Stenotrophomonas maltophilia isolate (Rahman et al., 2007) with Shigella dysenteriae strains and to identify the putative cellular components giving rise to this type of serological cross-reactivity.
MATERIALS AND METHODS
Bacterial strains: The test organism Stenotrophomonas maltophilia (RBSD4), was an environmental isolate and three clinical Shigella dysenteriae strains (S. dysenteriae type 2 (602), type 3 (C253) and type 4 (613)) were used as reference. All the organisms were obtained from the Environmental Microbiology Laboratory, Department of Microbiology, University of Dhaka, Bangladesh in 2009.
Biotyping of the test isolate Stenotrophomonas maltophilia: Biotyping of the test isolate S. maltophilia along with the reference Shigella dysenteriae strains were performed to reveal similarities or dissimilarities in their biochemical behavior.
Detection of Shigella-specific virulence genes (ipaH, virA and stx1) by PCR
Extraction of chromosomal DNA: Chromosomal DNA of the test isolate and the reference strains were extracted and purified according to the method described by Sambrook et al. (1989). Following this method, pellets from overnight fresh culture were collected by centrifugation at 10,000 rpm for 5 min and were resuspended in 100 μL of solution I containing 50 mM Tris-HCl buffer (pH: 7.5), 50 mM EDTA and 20% (w/v) sucrose. This was followed by addition of 300 μL of solution II containing 1% (w/v) SDS and 50 mM NaCl and 5 μL of proteinase K (20 mg mL-1) to each eppendorf. After gentle shaking, the pellet suspension was incubated at 55°C for 60 min in a water bath. DNA was extracted by using 400 μL phenol: chloroform: isoamylalcohol mixture (25:24:1). The and the top phase was recovered carefully into fresh tubes were mixed well, centrifuged for 10 min at 10,000 rpm eppendorf tubes. Double volume of ice-cold 100% ethanol followed by 1/10th volume of the 3 M sodium acetate (pH: 5.2) were added to the DNA solution, mixed well and then kept at -20°C overnight. DNA pellet collected by centrifugation at 10,000 rpm for 10 min was washed again with 70% ethanol and was finally dried in a desiccator under vacuum and resuspended in 50 μL TE buffer.
PCR: The PCR was performed in a 25 μL volume consisting of 0.5 μg of genomic DNA, 0.5 mM of each of the oligonucleotide primers (Table 1) for ipaH, virA and stx1, 2.5 μL of a 10xPCR reaction buffer (500 mM Tris-Cl, pH 8.9, 500 mM KCl and 25 mM MgCl2), 0.5 μL 10 mM dNTPs, 1.25 units of AmpliTaq DNA polymerase (Invitrogen, Life Technologies, USA) and an appropriate volume of sterile MilliQ water. Thermal cycling conditions were set for denaturation at 94°C for 1 min, annealing of primers at 55°C for 1 min 30 sec and primer extension at 72°C for 1 min 30 sec. Amplification was performed for 35 cycles, The expected sizes of the amplicons were ascertained by electrophoresis in 1.0% agarose gels (10 μL PCR product loaded) with an appropriate molecular size marker (1-kb DNA ladder; Sigma, USA).
Plasmid profile analysis: Plasmid extraction from the test isolates was carried out according to the procedure described by Kado and Liu (1981). Overnight fresh culture was centrifuged at 12,000 rpm for 7 min and pellets were resuspended in 100 μL autoclaved solution I (50 mM glucose, 25 mM Tris-HCl, 10 mM EDTA at 4°C and pH: 8.0) and kept at room temperature for 5 min. Freshly prepared 200 μL of solution II (0.2 N NaOH, 1% SDS in deionized water) was then added, mixed gently and kept in ice for 10 min. This was followed by addition of 150 μL of solution III (3 M potassium acetate, 5 M glacial acetic acid, pH: 4.8), mixed gently and kept in ice for 5 min. The suspension was centrifuged at 12,000 rpm for 5 min and 300 μL of supernatant was collected in fresh eppendorf tubes. Double volume of ice cold ethanol (95%) was added to the supernatant, mixed well and kept at room temperature for 10 min. Following centrifugation at 12,000 rpm for 5 min, the pellet was taken out and mixed with 1.0 mL ethanol (70%).
| Table 1: | Primers used for the detection of Shigella-specific genes (ipaH, virA and stx1) by polymerase chain reaction (PCR) |
 | |
This was centrifuged again at 12,000 rpm for 5 min. Finally, the collected pellet was dried and resuspended in 50 μL TE buffer and kept at 4°C.
Plasmid DNA was separated by horizontal electrophoresis in 1.0% agarose gel slab in Tris-acetate EDTA (TAE) buffer at room temperature at 50 volts for 3 h. The gel was stained with ethidium bromide and DNA bands were visualized using UV transilluminator (Gel Doc, Bio-Rad, USA).
Extraction of Outer Membrane Protein and Lipopolysaccharide (LPS): The outer membrane proteins were extracted from the isolates by the water extraction method described by Hale et al. (1985). Briefly, an isolated colony from a fresh culture in Nutrient Agar (NA) was inoculated into brain heart infusion broth and incubated at 37°C for 24 h in an orbital shaker. Following incubation, the whole culture media was harvested by centrifugation at 8,000 rpm for 15 min. The pellet was washed in normal saline and resuspended in 5.0 mL of sterilized deionized water and was shaken (100 oscillations per min) for 6 h at room temperature. The suspension was then centrifuged at 10,000 rpm for 20 min and the supernatant was collected and filtered through a Millipore filter having pore size of 0.45 μm. LPS was extracted using extraction kit (iNTRON Biotechnology, http://eng.intronbio.com/product/LPS.htm). Finally, 70 μL of 10 mM Tris-HCl buffer (pH: 8.0) was added to the LPS pellet and was dissolved by boiling for 1 min.
Western blot analysis: Outer Membrane Proteins (OMP) and LPS were fractionated separately by sodium-dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 15% polycrylamide gels following the procedure described by Laemmli (1970). Twelve microlitre of OMP and LPS were loaded. These were followed by Western blot analyses (Towbin and Gordon, 1984). LPS and OMP were translocated from the gel to the nitrocellulose membrane by overnight transfer in transfer buffer (1.8% glycine, 0.36% Tris, 25% methanol in deionized water). The membrane was then cut into strips, washed with PBS (Phosphate Buffer Saline), PBS-Tween20 (0.1%) and subjected to blocking solution (3% skim milk in PBS). Commercially available antisera specific to S. dysenteriae type 2, 3, 4 and 8 were used at a concentrations of 1:50 dilutions as primary antisera (Denka Seiken, Tokyo, Japan). Horse radish peroxidase (HRP)-conjugated antibody (anti-rabbit IgG; Sigma) at 1:2000 dilutions was used as secondary antibody. Diaminobenzoic acid (DAB) (0.1%) dissolved in 100 mM citrate buffer (2.94% Na-citrate in deionized water, pH 5.2) with 0.15% H2O2 was used as the substrate.
Proteolytic, hemolytic and hemagglutinating activity: Proteolytic activity was determined based on the ability to hydrolyze milk protein casein. Fresh cultures were inoculated in skim milk agar plate and incubated overnight at 37°C (Murray et al., 1999).
The hemolytic activities of the test S. maltophilia isolate and reference S. dysenteriae strains were determined by streaking aseptically on the blood agar plates. After incubation at 37°C for 18-20 h in aerobic and anaerobic jar incubator, the hemolysis zone and type under both the conditions were determined respectively (Atlas, 2006).
The hemagglutinating study was performed in microtiter plate. The chicken blood cells were washed thrice in 0.01 M cold PBS and finally, suspended to a concentration of 10% (v/v). Fifty microlitre of chicken blood cell suspension was added to each well. Bacterial suspension containing 1010 colony forming unit (cfu) mL-1 was subjected to two-fold serial dilution in 0.01 M PBS. Fifty microlitres of serially diluted test isolate and Shigella strains were then added to the wells and the microtiter plate was incubated at 22°C for 1 h. The highest dilution giving complete hemagglutination was recorded as the end point (Franzon and Manning, 1986).
Test of invasiveness: Sereny (keratoconjunctivitis) test was used as confirmatory test of invasiveness. Bacterial suspension adjusted to a concentration of 3×108 cfu mL-1 was used as inocula for Sereny test. Thirty microliter of this suspension from each organism was dispensed into the left eye of each guinea pig. Cell suspensions of Shigella flexneri 2a was used as positive control (Sereny, 1955).
Rabbit Ileal Loop (RIL) assay for enterotoxicity: Preparation of inocula was done following the method of De and Chatterje (1953). Both live cells and culture filtrates were used as inocula. Prepared inoculum cell suspension and culture filtrate (1.0 mL each) of the test isolate and reference strain together with positive (Vibrio cholerae 569B) and negative (PBS) controls were injected separately in the ligated loops of an Albino rabbit. The same method was repeated twice to check for reproducibility of results. The animals were sacrificed after 18 h by intravenous administration of 10% magnesium sulfate. Enterotoxicity was determined according to the fluid accumulation ratio suggested by De and Chatterje (1953).
Antibiotic susceptibility pattern: Susceptibility of the test isolate and the reference strains were determined by using the standardized agar-disc-diffusion method known as Kirby-Bauer (Barry et al., 1985). A total of seventeen different commercially available (Oxoid Limited, England) antibiotic discs were used in this study. The zone diameter for individual antimicrobial agent was then translated into sensitive, intermediate or resistant categories according to the interpretation table (Oxoid Limited, England).
RESULTS
Biotyping of the test isolate: The test isolate Stenotrophomonas maltophilia and the reference Shigella dysenteriae strains had many similar biochemical properties. But in contrast to the reference strains, the test isolate was unable to utilize any of the carbohydrates and possessed lysine decarboxylase activity (Table 2).
Detection of Shigella-specific virulence genes and plasmid profile analysis: The test isolate S. maltophilia did not harbor ipaH, virA or stx1 genes (Fig. 1, 2), whereas all the reference strains demonstrated ipaH and virA positive results and 421 and 215 bp amplified bands were observed in the agarose gel respectively. In addition, the reference strain S. dysenteriae type 3 (C253) was also stx1 positive and gave 384 bp amplified band.
Analysis of plasmid DNA by agarose gel electrophoresis revealed that the test isolate S. maltophilia had no plasmid. Only the reference strain S. dysenteriae type 4 (613) had large plasmid and other reference strains contained plasmid bands ranging between approximately 2.9 and 14 kb (Fig. 3).
LPS and OMP profile analysis: Western blot analysis of the LPS of the test isolate S. maltophilia did not reveal any cross-reactivity with S. dysenteriae type 8 specific antisera. This suggested that, the cross reactivity of this isolate was not due to its LPS. Any other outer membrane protein or surface protein might contribute to such type of serological cross-reactivity. Therefore, this assumption was confirmed by the OMP profile analysis (Fig. 4) which showed antigenic bands on the nitrocellulose membrane.
Proteolytic, hemolytic and hemagglutinating activity: The test isolate S. maltophilia gave clear zone of proteolysis on skim milk agar whereas all the reference Shigella dysenteriae strains demonstrated negative result.
In case of the test Stenotrophomonas maltophilia isolate, zones of α-hemolysis (greenish hue) were detected in blood agar plates incubated under aerobic as well as anaerobic conditions.
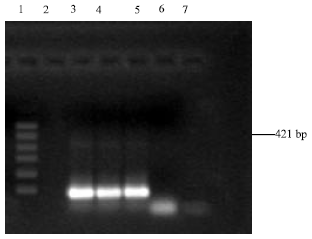 | |
| Fig. 1: | PCR analysis of the test S. maltophilia isolate for Shigella-specific virulence gene, ipaH. Products of a PCR assay for the ipaH gene are shown. Lane 1: 1 kb+ DNA ladder marker; Lane 3: S. dysenteriae type 2 (602); Lane 4: S. dysenteriae type 5 (613); Lane 5: S. dysenteriae type 3 (C253); Lane 6: S. maltophilia; Lane 7: Negative control |
| Table 2: | Biochemical characteristics and antibiotic susceptibility pattern of the environmental test isolate S. maltophilia along with the reference S. dysenteriae strains |
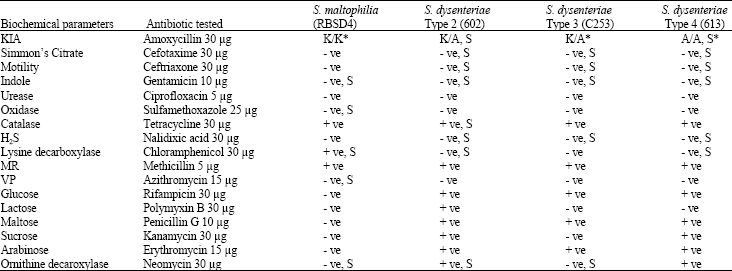 | |
| *K: Alkaline reaction, A: Acid reaction, S: Sensitive to antibiotic | |
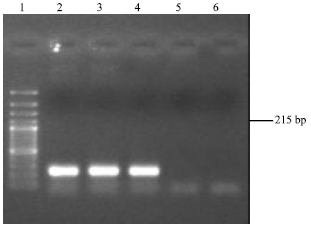 | |
| Fig. 2: | PCR analysis of the test S. maltophilia isolate for Shigella-specific virulence gene, virA. Products of a PCR assay for the virA gene are shown. Lane 1: 1 kb+ DNA ladder marker; Lane 2: S. dysenteriae type 2 (602); Lane 3: S. dysenteriae type 4 (613); Lane 4: S. dysenteriae type 3 (C253); Lane 5: S. maltophilia, Lane 6: Negative control |
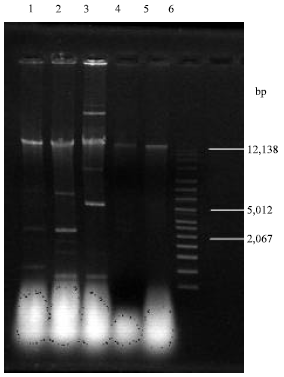 | |
| Fig. 3: | Plasmid profile analysis. Lane 1: S. dysenteriae type 3 (C253); Lane 2: S. dysenteriae type 2 (602); Lane 3: S. dysenteriae type 4 (613); Lane 5: S. maltophilia; Lane 6: Supercoiled DNA ladder |
But the reference strains (S. dysenteriae type 2, 3 and 4) did not show any hemolysis (gamma hemolysis) on blood agar plate incubated either in aerobic or in anaerobic condition.
 | |
| Fig. 4: | OMP profile of the test isolate S. maltophilia (RBSD4) along with three clinical S. dysenteriae strains. Lane 1: protein marker; Lane 2 and 3: test isolate S. maltophilia; Lane 4 and 5: S. dysenteriae type 2 (602); Lane 6 and 7: S. dysenteriae type 4 (613); Lane 8 and 9: S. dysenteriae type 3 (C253) |
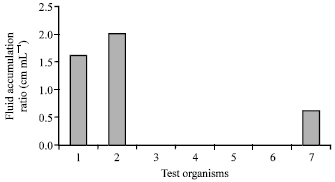 | |
| Fig. 5: | Bar chart showing the ratio of fluid accumulation (mL cm-1) by the test isolate, reference strains and positive control in RIL assay. Key: 1: V. cholerae 569B live cells (positive control); 2: V. cholerae 569B culture filtrate (positive control); 3: Test S. maltophilia live cells; 4: Test S. maltophilia culture filtrate; 5: S. dysenteriae type 2 (602); 6: S. dysenteriae type 3 (C253); 7: S. dysenteriae type 4 (613) live cells |
Neither the test isolate S. maltophilia (RBSD4) nor the reference strains (S. dysenteriae type 2, 3 and 4) exhibited any hemagglutinating activity in vitro.
Sereny (keratoconjunctivitis) assay: Sereny test revealed that with the test S. maltophilia did not cause any sign of inflammation in guinea pig eyes. Only those inoculated with the reference strain S. dysenteriae type 4 (613) and positive control S. flexneri 2a exhibited typical symptoms of keratoconjunctivitis.
Rabbit Ileal Loop (RIL) assay: Level of enterotoxicity was determined according to the ratio of fluid accumulation (mL) per unit (cm) length of ileal loop. Response is considered positive when the fluid accumulation reflects≥0.4 mL cm-1. Live cell and culture filtrate of the test S. maltophilia (RBSD4) did not cause any fluid accumulation. Enterotoxicity was evident in RIL only with the reference strain S. dysenteriae type 4 (613) and the positive control Vibrio cholerae 569B (Fig. 5).
Antibiotic susceptibility pattern of the test and reference strains: The test isolate S. maltophilia showed resistance to methicillin, erythromycin, amoxycillin, cefotaxime, ceftriaxone and penicillin G. It also showed intermediate resistance to antibiotics like tetracycline, nalidixic acid, rifampicin, polymyxin B and kanamycin. The test isolate was sensitive to gentamicin, ciprofloxacin, chloramphenicol and trimethoprim sulfamethoxazole (Table 2).
DISCUSSION
Shigellosis is one of the dreadful diarrheal diseases due to its invasive character, systemic manifestations, severe nutritional impact and tendency to recur over prolonged periods (Mata et al., 1970). In the present study, the virulent properties of serologically cross-reactive environmental Stenotrophomonas maltophilia were compared with reference Shigella dysenteriae strains. The serologically cross-reactive S. maltophilia isolate revealed many similarities and dissimilarities in their biochemical behavior. In contrast to the reference strains, the S. maltophilia isolate was unable to utilize carbohydrates, e.g., glucose, lactose, sucrose, maltose and arabinose. This finding is not in conformity to another report which showed that some clinical S. maltophilia strains were capable of utilizing glucose, lactose, sucrose, maltose, xylose and citrate (Travassos et al., 2004). Another distinguishing characteristic of the test isolate was that it possessed lysine decarboxylase activity while all the reference Shigella strains were lysine decarboxylase negative. Lysine decarboxylase (LDC) activity is present in ≈90% of E. coli strains (Edwards and Ewing, 1972) but all the strains of EIEC and Shigella sp. are LDC negative (Silva et al., 1980). When the gene for LDC, cadA was introduced into Shigella flexneri 2a, virulence became attenuated and enterotoxin activity was inhibited significantly. The enterotoxin inhibitor was identified as cadaverine, a product of the reaction catalyzed by LDC. Comparison between the S. flexneri 2a and E. coli K-12 genomes in the region of cadA revealed a large deletion in Shigella. Representative strains of Shigella sp. and enteroinvasive E. coli displayed similar deletions of cadA (Maurelli et al., 1998). This study is consistent with the present findings with the test S. maltophilia isolate which was lysine decarboxylase positive but lacked enterotoxic property.
Presence of Shigella-specific virulence genes have been reported in closely related Escherichia coli virotypes (Pass et al., 2000). In this study, serologically cross-reactive environmental S. maltophilia isolate demonstrated PCR negative results for Shigella-specific virulence genes like ipaH, virA and stx1 indicating that this organism did not possess any invasive or enterotoxigenic property. Absence of Shigella-specific virulence genes is a prerequisite for a vaccine candidate against shigellosis (DuPont et al., 1972). This serologically cross-reactive environmental S. maltophilia isolate fulfils the criterion and may open up the scope for development of shigellosis vaccine based on live cells.
Invasive organisms like E. coli (EIEC) and Shigella sp., cause a rapid keratoconjunctivitis when placed on the conjunctiva of the guinea pig eye (known as Sereny test). Manifestations of keratoconjunctivitis include white or yellowish discharge from the eye, crustiness around the eye, redness or swelling in and around the eye and behavior such as pawing at the eyes or sneezing. Sereny test-positive isolates carry large (usually 140-MDa) plasmid responsible for this property. In the present study, the test isolate S. maltophilia gave negative response in the Sereny test indicating that it was noninvasive. Only the reference strain of S. dysenteriae type 4 (613) produced typical symptoms of keratoconjunctivitis in inoculated guinea pig eyes. This result is consistent with the plasmid profile analysis which showed the presence of a large virulence plasmid only in S. dysenteriae type 4 (613) and its absence in all the other reference strains including the test isolate.
The serological cross-reactivity is mostly due to the heat stable LPS antigens or common outer membrane protein antigens (Peterfi et al., 2007). In the present study, Western blot analysis revealed that the cross-reactivity with S. maltophilia isolate was not due to its LPS component. As many protein components in the outer membrane are also shared among different Gram negative bacteria, the cross-reaction observed in the test isolate might be due to any outer membrane protein(s). This assumption was eventually confirmed by Western blot analysis involving outer membrane protein (OMP) of the bacterial isolate. The environmental S. maltophilia isolate yielding a high cross-reactivity with Shigella antisera represents potential for development of a shigellosis vaccine.
The ability of organisms to produce enterotoxins and hence their diarrhoeagenic potential was tested using the rabbit ileal loop assay. The test isolate S. maltophilia live cells and its culture filtrate did not cause any fluid accumulation indicating that it was incapable of producing enterotoxin. Absence of enterotoxic property is consistent with the finding that it did not possess any toxin encoding gene.
Active extracellular protease was secreted by the test isolate. Some clinical S. maltophilia isolate also produce alkaline serine protease detected as a major secretion product (Windhorst et al., 2002). Stenotrophomonas maltophilia G2 isolated from a soil sample producing extracellular serine protease have been documented in a separate study (Huang et al., 2009). The roles played by protease in this environmental S. maltophilia isolate remains to be explored. The test S. maltophilia isolate exhibited alpha hemolysis on blood agar plate indicating that it was capable of partially degrading hemoglobin, giving rise to the typical greenish hue around bacterial growth. An earlier study which reported clinical S. maltophilia presenting a cell-free hemolytic activity similar to the 'hot-cold' hemolysins (Figueiredo et al., 2006) is in conformity to the present findings. Hemolysins may be channel-forming proteins or phospholipases or lecithinases that destroy red blood cells and other cells by lysis (Figueiredo et al., 2006). No hemagglutinating activity in vitro was exhibited by the test isolate and the reference strains. This indicates the absence of specific hemagglutinin on the bacterial cell surface which interacts with the chicken red blood cell membrane component.
Resistance to β-lactam antibiotics was confirmed previously in a study reporting approximately 200 kb plasmid purified from clinical isolates of S. maltophilia (Avison et al., 2001). Transformation of a 5.6 kb plasmid, designated pTHB, derived from a S. maltophilia isolate into Escherichia coli K-12 HB101 resulted in the expression of resistance to the penicillin and to cefazolin (Kelly et al., 1995). However, the occurrence of multiple antibiotic resistance devoid of any plasmid in the test isolate indicates that these resistance genes might be chromosome mediated. This type of antibiotic resistance might be due to the occurrence of certain genes like Smqnr, a new chromosome-borne quinolone resistance gene, in S. maltophilia (Shimizu et al., 2008).
The overall findings of this study suggest the S. maltophilia isolate to be noninvasive and nonenterotoxic. Multiple antibiotic resistance and production of hemolysin and extracellular protease are some prominent features of the test isolate. Routine procedures to identify S. dysenteriae must incorporate virulence tests in addition to culture and serology in order to rule out any possibility of misdiagnosis. Finally, further investigations are necessary to fully characterize the virulent properties of this environmental isolate and its potential as a vaccine candidate against shigellosis.
ACKNOWLEDGMENT
This study was supported by The Norwegian Programme for Development, Research and Higher Education (NUFU), grant No NUFUPRO-2007/10063.
REFERENCES
- Avison, M.B., C.S. Higgins, C.J.V. Eldreich, P.M. Bennett and T.R. Walsh, 2001. Plasmid location and molecular heterogeneity of the L1 and L2 β lactamase genes of Stenotrophomonas maltophilia. Antimicrob. Agents Chemother., 45: 413-419.
CrossRef - Barry, A.L., C. Thornsberry, R.N. Jones and T.L. Gavan, 1985. Aztreonam: Antibacterial activity, β-lactamase stability, and interpretive standards and quality control guidelines for disk-diffusion susceptibility tests. Rev. Infect. Dis., 7: 594-604.
Direct Link - De, D.N. and S.N. Chatterje, 1953. An experimental study of the mechanism of action of Vibrio cholerae on the intestinal mucous membranes. J. Pathol. Bacteriol., 66: 559-562.
Direct Link - DuPont, H.L., R.B. Hornick, M.J. Snyder, J.P. Libonati, S.B. Formal and E.J. Gangarosa, 1972. Immunity in shigellosis. I. Response of man to attenuated strains of Shigella. J. Infect. Dis., 125: 5-11.
Direct Link - Evins, G.M., L.L. Gheesling and R.V. Tauxe, 1988. Quality of commercially produced Shigella serogrouping and serotyping antisera. J. Clin. Microbiol., 26: 438-442.
Direct Link - Franzon, V.L. and P.A. Manning, 1986. Molecular cloning and expression in Escherichia coli K-12 of the gene for a hemagglutinin from Vibrio cholerae. Infection Immunity, 52: 279-284.
Direct Link - Figueiredo, P.M.S., M.T. Furumura, A.M. Santos, A.C.T. Sousa, D.J. Kota, C.E. Levy and T. Yano, 2006. Cytotoxic activity of clinical Stenotrophomonas maltophilia. Lett. Applied Microbiol., 43: 443-449.
PubMed - Grubor, N.M., D.W. Armstrong and R. Jankowiak, 2006. Flow-through partial-filling affinity capillary electrophoresis using a cross-reactive antibody for enantiomeric separations. Electrophoresis, 27: 1078-1083.
PubMed - Hale, T.L., E.V. Oaks and S.B. Formal, 1985. Identification and antigenic characterization of virulence-associated, plasmid encoded proteins of Shigella spp. and enteroinvasive Escherichia coli. Infect. Immun., 50: 620-629.
PubMed - Houpikian, P. and D. Raoult, 2003. Western immunoblotting for Bartonella endocarditis. Clin. Diagn Lab. Immunol., 10: 95-102.
PubMed - Huang, X., J. Liu, J. Ding, Q. He, R. Xiong and K. Zhang, 2009. The investigation of nematocidal activity in Stenotrophomonas maltophilia G2 and characterization of a novel virulence serine protease. Can. J. Microbiol., 55: 934-942.
PubMed - Jin, L.Q., L.W. Li, S.Q. Wang, F.H. Chao, X.W. Wang and Z.Q. Yuan, 2005. Detection and identification of intestinal pathogenic bacteria by hybridization to oligonucleotide microarrays. World J. Gastroenterol., 11: 7615-7619.
PubMed - Kado, C.I. and S.T. Liu, 1981. Rapid procedure for detection and isolation of large and small plasmids. J. Bacteriol., 145: 1365-1373.
Direct Link - Kelly, M.D., J.E. Mortensen, B.A. Konkle and T.L. Stull, 1995. A plasmid mediating production of a beta-lactamase by Stenotrophomonas maltophilia. Curr. Therapeutic Res., 56: 152-162.
CrossRef - Khan, A., R.K. Nandi, S.C. Das, T. Ramamurthy, J. Khanam and T. Shimizu et al., 2003. Environmental isolates of Citrobacter braakii that agglutinate with Escherichia coli O157 antiserum but do not possess the genes responsible for the biosynthesis of O157 somatic antigen. Epidemiol. Infect., 130: 179-186.
Direct Link - Laemmli, U.K., 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227: 680-685.
CrossRefDirect Link - Lefebvre, J., F. Gosselin, J. Ismail, M. Lorange, M. Lior and D. Woodward, 1995. Evaluation of commercial antisera for Shigella serotyping. J. Clin. Microbiol., 33: 1997-2001.
Direct Link - Mata, L.J., E.J. Gangarosa, A. Caceres, D.R. Perera and M.L. Mejicanos, 1970. Epidemic shiga bacillus dysentery in central America. I. Etiological investigation in Guatemala, 1969. J. Infect. Dis., 122: 170-180.
Direct Link - Maurelli, A.T., R.E. Fernandez, C.A. Bloch, C.K. Rode and A. Fasano, 1998. Black holes and bacterial pathogenicity: A large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. USA., 95: 3943-3948.
Direct Link - Paton, A.W. and J.C. Paton, 1998. Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111 and rfbO157. J. Clin. Microbiol., 36: 598-602.
PubMedDirect Link - Pass, M.A., R. Odedra and R.M. Batt, 2000. Multiplex PCRs for identification of Escherichia coli virulence genes. J. Clin. Microbiol., 38: 2001-2004.
Direct Link - Peterfi, Z., I. Kustos, F. Kilar and B. Kocsis, 2007. Microfluidic chip analysis of outer membrane proteins responsible for serological cross-reaction between three Gram-negative bacteria: Proteus morganii O34, Escherichia coli O111 and Salmonella Adelaide O35. J. Chromato A, 1155: 214-217.
PubMed - Rahman, Z.R., M. Sultana, S.I. Khan and N.K. Birkeland, 2007. Serological cross-reactivity of environmental isolates of Enterobacter, Escherichia, Stenotrophomonas and Aerococcus with Shigella spp.-specific antisera. Curr. Microbiol., 54: 63-67.
CrossRef - Sereny, B., 1955. Experimental Shigella keratoconjunctivitis. Acta Microbiol. Acad. Sci. Hung., 2: 293-296.
PubMed - Silva, R.M., R.F. Toledo and L.R. Trabulsi, 1980. Biochemical and cultural characteristics of invasive Escherichia coli. J. Clin. Microbiol., 11: 441-444.
Direct Link - Sambrook, J., E.F. Fritsch and T.A. Maniatis, 1989. Molecular Cloning: A Laboratory Manual. 1st Edn., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Direct Link - Seidlein, V., D.R. Kim, M. Ali, H. Lee and X. Wang et al., 2006. Multicentre study of Shigella diarrhoea in six Asian countries: Disease Burden. Clin. Manifestations Microbiol. PLoS Med., 3: e353-e353.
PubMed - Shimizu, K., K. Kikuchi, T. Sasaki, N. Takahashi, M. Ohtsuka, Y. Ono and K. Hiramatsu, 2008. Smqnr, a new chromosome-carried quinolone resistance gene in Stenotrophomonas maltophilia. Antimicrob Agents Chemother., 52: 3823-3825.
CrossRef - Towbin, H. and J. Gordon, 1984. Immunoblotting and dot immunoblotting�current status and outlook. J. Immunol. Methods, 72: 313-340.
PubMed - Travassos, L.H., M.N. Pinheiro, F.S. Coelho, J.L.M. Sampaio, V.L.C. Merquior and E.A. Marques, 2004. Phenotypic properties, drug susceptibility and genetic relatedness of Stenotrophomonas maltophilia clinical strains from seven hospitals in Rio de Janeiro, Brazil. J. Applied Microbiol., 96: 1143-1150.
PubMed - Venkatesan, M.M., J.M. Buysse and D.J. Kopecko, 1989. Use of Shigella flexneri ipaC and ipaH gene sequences for the general identification of Shigella spp. and enteroinvasive Escherichia coli. J. Clin. Microbiol., 27: 2687-2691.
Direct Link - Windhorst, S., E. Frank, D.N. Georgieva, N. Genov, F. Buck, P. Borowski and W. Weber, 2002. The major extracellular protease of the nosocomial pathogen Stenotrophomonas maltophilia. J. Biological Chem., 277: 11042-11049.
CrossRef