
J. A. Qasem
Department of Applied Medical Sciences, College of Health Sciences, Public Authority for Applied Education and Training, Kuwait
S. Al-Zenki
Department of Biotechnology, Kuwait Institute for Scientific Research, Kuwait
A. Al-Marzouk
Department of Aquaculture, Fisheries and Marine Environment, Kuwait Institute for Scientific Research, Kuwait
Pakistan Journal of Biological Sciences
Year: 2010 | Volume: 13 | Issue: 1 | Page No.: 9-15







ABSTRACT
This study was undertaken to apply 16S rRNA sequence and Cellular Fatty Acid (CFA) composition analysis techniques for the identification and characterization of seven individual bacteria isolates obtained from seven infected fish samples. All samples were cultured on brain heart infusion agar. All the seven isolates were Gram positive and were identified as Streptococcus sp. The 16S rRNA sequencing method yielded about 1500 bps for each strain where upon the sequence was compared to nucleotide data in Gene Bank using BLASTN 2.2.1 sequence alignment from NCBI for the nucleotide comparison. The 16S rRNA gene sequence for all the seven samples had no sequence variation between the isolates and gave a 100% similarity to plus, plus strand with Streptococcus agalactiae strain A909 Accession number NC_007432.1 and S. agalactiae strain H36B (Accession number AAJS01000007). Also the 16S rRNA sequence showed a high (92-93%) similarity between S. agalactiae and S. equi, S. suis and S. uberis. All strains appeared to be nearly identical to each other after CFA analysis using Library Generation System (LGS) software (MIDI) and were consistent to that of S. agalactiae ATCC 12386, the CFA analysis not only confirmed the results of 16S rRNA sequence but also indicated a possibility of single source of infection. Despite their accuracy to identify the poorly described, rarely isolated, or phenotypically aberrant strains, 16S rRNA gene sequence analysis and CFA analysis lacks widespread use beyond the large and reference laboratories because of technical and cost considerations.
PDF Abstract XML References Citation
How to cite this article
J. A. Qasem, S. Al-Zenki and A. Al-Marzouk, 2010. Identification and Characterization of Streptococcus agalactiae Isolates using 16S rRNA Sequencing and Cellular Fatty Acid Composition Analysis. Pakistan Journal of Biological Sciences, 13: 9-15.
DOI: 10.3923/pjbs.2010.9.15
URL: https://scialert.net/abstract/?doi=pjbs.2010.9.15
DOI: 10.3923/pjbs.2010.9.15
URL: https://scialert.net/abstract/?doi=pjbs.2010.9.15
INTRODUCTION
A disease breakout in wild mullet was observed in Kuwait Bay from August to September 2001 (Evans et al., 2002). Microbiological investigations strongly suggested streptococcal infection as major cause for fish mortality in Kuwait aqua environment (Evans et al., 2002; Olivares-Fuster et al., 2008). The objective of this work was to use of 16S rRNA sequencing and Cellular Fatty Acid (CFA) composition analysis for species identification of the isolates.
The analysis of cellular fatty acids for bacteria identification has been used for many years. The availability of high resolution gas chromatography coupled with a computer data base has allowed this method to become widely accepted (Welch, 1991).
Bacteria grown under controlled conditions exhibit free fatty acid profiles which can be used for their identification. These qualitative and quantitative differences in cellular fatty acids profiles can differentiate genus, species and sub-species of the bacteria. Whole cell bacteria extracts contain straight and branched fatty acids with 9 to 20 carbon lengths (saturated or unsaturated). The presence or absence of the over 300 different compounds along with their quantitative composition yields a wealth of taxonomic information which allows bacterial identification to species. (Lechevalier, 1982; Welch, 1991).
Alternatively, the use of chemical analysis of bacterial components, i.e., lipids, polysaccharides, proteins and nucleic acids, has increasingly been applied to bacterial taxonomy (Moss et al., 1980; Moss, 1981). Bacterial lipid determination, particularly cellular fatty acid analysis by gas-liquid chromatography, has long been recognized as a valuable chemotaxonomic tool for the classification and identification of nonfermentative gram-negative bacteria (Dees and Moss, 1975; Lechevalier, 1982; Moore et al., 1987; Moss, 1981; Veys et al., 1989). Compared with conventional and other chemical identification methods, other advantages of cellular fatty acid analysis include its relative speed and simplicity and the lack of the need for re-cultivation of the organism after initial growth or specialized and expensive reagents (Miller, 1982). Bacterial fatty acids, unlike many phenotypic characteristics, are genetically highly conserved owing to their essential role in cell structure and function. In addition, technical advances in recent years through the development of fused-silica capillary columns, automatic injection systems, digital integrators and standardized calibrators have increased the applicability of this technology to the clinical microbiology laboratory (Moss et al., 1980; Lehtonen et al., 1996). Despite these improvements, cellular fatty acid analysis and subsequent bacterial identification have relied on manual fatty acid identification and chromatogram interpretation, which require considerable time and expertise on the part of the investigator. Recently, an automated gas-liquid chromatography system with a computer interface and software for the identification of bacteria on the basis of cellular fatty acid composition has been introduced. In this study we used two advanced methods for microbial identification based on the 16S rRNA gene sequence and the cellular fatty acid content for the identification of non-fermentative Gram-positive bacteria.
We report here the usefulness of CFA composition and 16S rRNA sequence as methods to identify and distinguish the Streptococcus on the species level isolated from environmental samples.
MATERIAL AND METHODS
Bacterial isolates and culture conditions: A total of 7 bacterial isolates from infected fish tissues were used in this study. The isolates were derived mainly from Mullet (Liza klunzingeri). Streptococcus sp., used in this study were isolated from the moribund fish in Kuwait Bay during outbreak that occurred in Kuwait bay 2001-2002 the samples were kept frozen at -80°C.
Isolates were provided by the Mariculture and Fisheries Department (MFD) at the Kuwait Institute for Scientific Research (KISR).
Working cultures were maintained on brain heart infusion agar (BHIA, Oxoid). When required, the culture was transferred from BHIA plates to brain heart infusion broth (BHIB, Oxoid) followed by appropriate incubation for 18-24 h at 37°C (Al-Marzouk et al., 2005). All cultures were maintained at 4°C in tubes with BHIB partially solidified with 0.8% agar. Purity was checked by plating on BHIA with 0.5% lactose.
DNA extraction and PCR amplification: The bacterial cells were harvested from 5 mL brain-heart infusion broth (109 cfu mL-1), re-suspended in 100 μL TE; total genomic DNA from the entire streptococcus isolates was purified by using a commercial kit (Promega MD, USA).
16S rRNA sequencing and analysis: Positive samples identified through bacteriology and PCR methods (Qasem et al., 2008) were selected from each fish species and were tested for species identity using 16S rRNA sequencing method which was further analyzed by Basic Local Alignment Search Tool for Nucleotides (BLASTN), National Center for Biotechnology Information (NCBI), USA (Altschul et al, 1990). The gene sequencing was done in National Microbiological Laboratory Health Canada using improved method developed by Bernard et al. (2002), it is based on Edwards et al. (1989). This method is similar to many other described in recent literature, yielding about 1500 bps for each strain whereupon the sequence is compared to 16S data in Genbank. Establishing the closest known relative was done by performing database search of GenBank with the Blast program. The phylogenetic tree was constructed based on Fast Minimum Evolution (FastME) (Desper and Gascuel 2002, 2006), the phylogenetic and molecular evolutionary analysis were conducted using Molecular Evolutionary Genetic Analysis (MEGA) Version 4.0. (Tamura et al., 2007).
Cellular fatty acid analysis: All strains were grown on 5% sheep blood agar with Columbia base for 24 h, 35-37°C, 5% CO2 prior to harvest. Each strain was extracted and processed as described by Bernard et al. (1991), using Version 3.9 or version 4.0 of the MIDI method aerobe (MIDI), New-ark, Del.). Cellular fatty acid composition analysis was done in National Microbiological Laboratory Health Canada And library generation system software (MIDI) were used to make entries for an in-house library. Identification were made based on a strain’s having a cellular fatty acid composition and products of fermentation consistent with species of the genus S. agalactiae ATCC 12386 (Aguggini et al., 1966). Values for fatty acids observed for each isolate represented a percentage of the total fatty acids for that isolate and were rounded off to the nearest whole percent. The entries consisted of arithmetic means of observed fatty acids for all isolates tested.
RESULTS AND DISCUSSION
The fatty acid composition of the seven presumptive Streptococcus sp., were determined, the CFA composition of the bacterial isolates studied consisted primarily of straight-chained saturated and monounsaturated fatty acids (Table 1).
| Table 1: | Cellular Fatty Acid (CFA) profile generated from fatty acids extracts of seven individual fish isolates belonging to Streptococcus sp. and an ATCC 12386 (reference strain used in these studies). The table represents the mean percent composition, standard deviation, and frequency percent of cellular fatty acids |
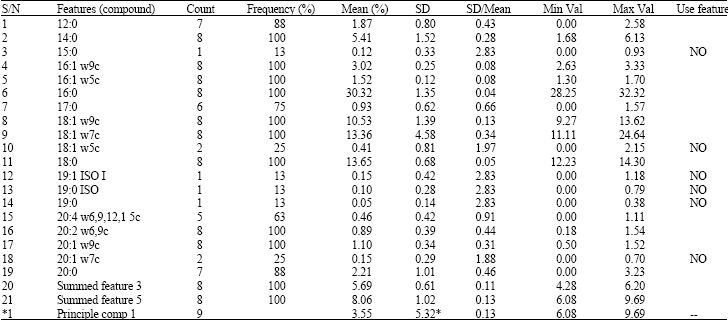 | |
*For the fatty acid composition the number to the left of the colon is the number of carbon atoms and the number to the right is the number of double bonds. *The portion of the double bound can be located by counting methyl w end of the carbon *chain A cis isomer is indicated by the suffix c. *SD: Stander deviation. *NO: Due to low frequency of the FA in the tested isolate it was excluded from final analysis | |
Six of the 21 different fatty acid types were excluded from the final data analysis since these were detected in 25% or less of the isolates (n1 or 2/7). The frequency and mean percentages of the other fatty acid methyl esters (FAMEs) are shown in Table 1. The mean values were calculated by using FAMEs data for the tested isolates, regardless of the fact that every fatty acid was not detected in all of the isolates. All seven isolates produced two summed features, the summed features represent groups of two or three fatty acids which could not be separated by gas-liquid chromatography with MIDI system, the summed feature 3 (16:1 w7c/15iso 2OH and/or 15:0 ISO 2OH/16: 1w7c) and summed feature 5 (18:2 w6, 9c/18:0 ANTE and/or 18:0 ANTE/18:2 w6, 9c) were found in all the isolates.
The Cellular fatty acid Identification were made based on a strain’s having cellular fatty acid composition and products of fermentation consistent with species of the genus Streptococcus agalactiae. Averaged fish isolate data plus ATCC 12386 gave rise to, using principle component analysis, a value of 5.32, (Table 1) whereas the same data with just fish isolates averaged 1.34. In analysis of data an averaged values of <10 suggests that the strains studied are members of the same taxon group. However, the lower the number, (such as found for fish isolates alone), the more the strains are considered as being like colonel. All strains appeared to be nearly identical to each other after CFA analysis using Library Generation System (LGS) software (MIDI) and were consistent to that of S. agalactiae ATCC 12386 (reference strain used in these studies) (Table 1).
To ensure 16S sequencing was performed on pure culture and to obtain classic bacteriological profiles, a number of accepted bacteriological tests (i.e., growth on differential media, biochemical tests, serological tests etc.) were executed (data not shown). The seven bacterial isolates were confirmed to be pure strains and were classified accordingly.
A significantly high degree of identity between the seven test isolates (100% similarity) with respect to 1,300 to 1,500 bp of 16 SrRNA gene sequence was found. The consensus sequence determined for the S. agalactiae 16S rRNA was examined for sequence homology with those of the prokaryotes by using National Centre for Biotechnology Information (NCBI), Gene Bank BLASTN function. The sequence alignment using BLASTN 2.2.1 for the comparison up to 1500 bp of the 16SrRNA gene sequence gave a high homology 100% to plus, plus strand with Streptococcus agalactiae strain A909 (ATCC 27591) (Accession number NC_007432.1) and S. agalactiae strain H36B (ATCC 12401; NCTC 8187). (Accession number AAJS01000007). The sequence alignment with sequence derived from validated S. agalactiae species (derived from GenBank) indicates the genus and species to be Streptococcus agalactiae (Table 2, Fig. 1).
Subsequently, an evolutionary distance matrix was then generated from the nucleotide sequences in the data set and a phylogenetic tree was constructed based on FastME algorithm using MEGA software. It clearly appears that all the seven isolates are affiliated with the species agalactiae.
| Table 2: | Similarity percentage of the top twenty microorganisms with the highest similarity percentage to the test isolates 16S rRNA sequence |
 | |
According to the phylogenetic tree, (Fig. 1) the species suis, equi, dysaglactiae, pyogenes, uberis, thermophilis and pneumoniae had a similarity of 90% or more with the test isolates. The most closely related species was S. suis and S. equi with similarity of 92 and 93 % to the test isolates (Table 2, Fig. 1).
Conventional and molecular identification methods were used to investigate the isolates of the different organs from the different fish were S. agalactiae. The biochemical characterization of the seven isolates supporting the identity of the isolated bacteria as a β-hemolytic group B Streptococcus, this was also confirmed by other work done by Al-Marzouk et al. (2005) and Evans et al. (2002, 2008) due to high similarity between S. iniae, Lactococcus graviae, S. dificile, S. shiloi with S. agalactiae. We established the species confirmation using two other molecular methods namely 16S rRNA gene sequencing method and CFA composition analysis results.
Limited information was available on the epidemiology of Kuwaiti S. agalactiae isolates recovered from different fish species. To our knowledge, only three studies using DNA-based techniques have been carried out with a large collection of field isolates of fish and sewage origin in Kuwait (Qasem et al., 2008; Olivares-Fuster et al., 2008; Duremdez et al., 2004).
In the present study, at least seven isolates with an identical 16SrRNA and CFA pattern were found and sequenced. This fact might suggest that, in some isolates, there may be a common source of S. agalactiae in different infected fish from the same region (Welch, 1991). No sequence variation was observed for fish isolates originating from the different fish species, except for those from same species (Mullet). This result is consistent with a previous report of Olivares-Fuster et al. (2008), who analyzed S. agalactiae of fish origin by single standard conformation polymorphism (SSCP). They reported the Kuwaiti isolates as one genotype group while the other clustered of non-Kuwaiti isolates (USA, Brazil and Honduras) indicating correlation between geographical origin of genotypes was observed (Olivares-Fuster et al., 2008).
The traditional identification of bacteria on the basis of phenotypic characteristics is generally not accurate as identification based on genotypic methods. Comparison of the bacterial 16S rRNA gene sequence has emerged as a preferred genetic technique. The 16S rRNA gene sequence analysis can better identify poorly described, rarely isolated, or phenotypically aberrant strains (Woese et al., 1985). The 16S rRNA gene sequencing was conformed the species identity of the 7 fish isolates to be S. agalactiae. The sequencing results gave further support to the biochemical and PCR as to the identity of the isolates being S. agalactiae. Despite its accuracy, 16S rRNA gene sequence analysis lacks widespread use beyond the large and reference laboratories because of technical and cost considerations (Edwards et al., 1989).
The method used for the characterization was 16S rRNA sequencing, which is genomic approach to the classification of bacteria based upon the 16S ribosomal RNA gene (Lane et al., 1985). This gene is both ubiquitous and highly conserved among prokaryotic organisms, making it a useful primer site for gene amplification by PCR (Lin and Schwarz, 2003). Results emphasized the need to take a polyphasic approach when isolating and identifying bacterial isolates, particularly when they are to be used in future pathogenicity studies.
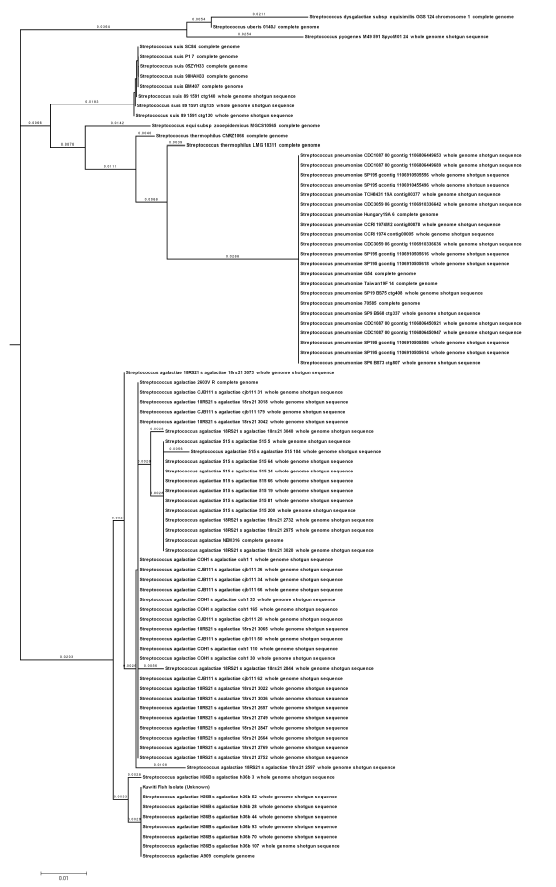 | |
| Fig 1: | Polygenetic tree based on Fast Minimum Evolution (FastME) after alignment of 1500 bp from 16 S rRNA sequences of the Kuwaiti isolates bacteria from diseased fish, with maximum sequence difference of 0.1 created using MEGA V 4.0 software |
The present study genetically characterized seven bacterial isolates derived from infected fish and compared these results against bacteriological and metabolic identification techniques to assess the need for a polyphasic approach when identifying bacteria from environmental and fish samples. As bacteriological and metabolic data showed discrepancies across different phenotypic analyses (Al-Marzouk et al., 2005), present results suggest the need to incorporate 16S rRNA analysis to identify the strains.
In conclusion the bacterial isolates of fish are identical or similar with high homology; this might suggest that the bacteria that infected the Mullet originated from the same source. This is the first work from fish kill in Kuwait using CFA technique to identify and characterize Streptococcus agalactiae from Kuwait. It is interesting that, by phylogenetic analysis of the 16S rRNA gene sequence, S. agalactiae; S. suis, S. uberis and S. equi were most closely related (92 to 93% identity) with each other but at times are difficult to differentiate even after using a polyphasic approach, for phenotypically aberrant strains (Evans et al., 2008). Also, the conclusion derived from CFA analysis were consistent with those derived from morphological, cultured and physical characteristic (Welch, 1991). We conclude that the utility of CFA testing as an in vitro procedure that can help to verify or to refine the identity of species of viable bacteria present in environmental samples.
ACKNOWLEDGMENTS
We thank Mr Schder C. Solman and Mss Salwa A. Al-Mouqati for there excellent technical assistance. And we also would like to thank Kuwait Institute for Scientific Research for partial funding of this work (FB019K).
REFERENCES
- Aguggini, G., L. Guallini and C. Gandini, 1966. On the fatty acid composition of lipids extracted from S. agalactiae. Arch. Vet. Ital., 17: 59-67.
PubMed - Al-Marzouk, A., R. Duremdez, K. Yuasa, S. Al-Zenki, H. Al-Gharabally and B. Munday, 2005. Fish Kill of Mullet Liza klunzingeri in Kuwait Bay: The role of Streptococcus agalactiae and the Influence of Temperature. In: Diseases in Asian Aquaculture V. Fish Health Section, Walker, P., R. Lester and M.G. Bondad-Reantaso (Eds.). Asian Fisheries Society, Manila, ISBN: 974-7313-64-2, pp: 143-153.
- Altschul, S.F., W. Gish, W. Miller, E.W. Myers and D.J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol., 215: 403-410.
CrossRefPubMedDirect Link - Bernard, K.A., M. Bellefeuille and E.P. Ewan, 1991. Cellular fatty acid composition as an adjunct to the identification of asporogenous, aerobic gram-positive rods. J. Clin. Microbiol., 29: 83-89.
PubMed - Bernard, K.A., C. Munro, D. Wiebe and E. Ongsansoy, 2002. Characteristics of rare or recently described corynebacterium species recovered from human clinical material in Canada. J. Clin. Microbiol., 40: 4375-4381.
PubMed - Dees, S.B. and C.W. Moss, 1975. Cellular fatty acids of Alcaligenes and Pseudomonas species isolated from clinical specimens. J. Clin. Microbiol., 1: 414-419.
PubMedDirect Link - Desper, R. and O. Gascuel, 2006. Getting a tree fast: Neighbor Joining, FastME and distance-based methods. Curr. Protoc. Bioinformatics.
CrossRefDirect Link - Edwards, U., T. Rogall, H. Blocker, M. Emde and E.C. Bottger, 1989. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucl. Acids Res., 17: 7843-7853.
CrossRefPubMedDirect Link - Evans, J.J., P.H. Klesius, P.M. Gilbert, C. Shoemaker and M. Al Sarawi et al., 2002. Characterization of β-haemolytic group B Streptococcus agalactiae in cultured seabream, Sparus auratus L. and wild mullet, Liza klunzingeri (Day), in Kuwait. J. Fish Dis., 25: 505-513.
CrossRefDirect Link - Evans, J.J., J.F. Bohnsack, P.H. Klesius, A.A. Whiting, J.C. Garcia, C.A. Shoemaker and S. Takahashi, 2008. Phylogenetic relationships among Streptococcus agalactiae isolated from piscine, dolphin, bovine and human sources: A dolphin and piscine lineage associated with a fish epidemic in Kuwait is also associated with human neonatal infections in Japan. J. Med. Microbiol., 57: 1369-1376.
PubMed - Lane, D.J., B. Pace, G.J. Olsen, D.A. Stahl, M.L. Sogin and N.R. Pace, 1985. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. USA., 82: 6955-6959.
PubMedDirect Link - Lehtonen, L., R. Peltonen and E. Eerola, 1996. Computerised gas-liquid chromatography of bacterial cellular fatty acids in analysis of bacterial mixtures. J. Microbiol. Methods, 25: 317-327.
CrossRef - Lin, M. and J.R. Schwarz, 2003. Seasonal shifts in population structure of Vibrio vulnificus in an estuarine environment as revealed by partial 16S ribosomal DNA sequencing. FEMS Microbiol. Ecol., 45: 23-27.
PubMed - Miller, L.T., 1982. Single derivatization method for routine analysis of bacterial whole-cell fatty acid methyl esters, including hydroxy acids. J. Clin. Microbiol., 16: 584-586.
PubMedDirect Link - Moore, C.J., H. Mawhinney and P.J. Blackall, 1987. Differentiation of Bordetella avium and related species by cellular fatty acid analysis. J. Clin. Microbiol., 25: 1059-1062.
PubMedDirect Link - Moss, C.W., S.B. Dees and G.O. Guerrant, 1980. Gas-liquid chromatograph y, of bacterial fatty acids with a fused-silica capillary column. J. Clin. Microbiol., 12: 127-130.
PubMedDirect Link - Qasem, j.A., S. Al-Zinki, S. Al-Mouqati, S. Al-Amad, A. Al-Marzouk and F. Al-Sharifi, 2008. Molecular investigation of Streptococcus agalactiae isolates from environmental samples and fish specimens during a massive fish kill in kuwait bay. Pak. J. Biol. Sci., 11: 2500-2504.
CrossRefPubMedDirect Link - Tamura, K., J. Dudley, M. Nei and S. Kumar, 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol., 24: 1596-1599.
CrossRefPubMedDirect Link - Veys, A., W. Callewaert, E. Waelkens and K.V.D. Abbeele, 1989. Application of gas-liquid chromatography to the routine identification of nonfermenting gram-negative bacteria in clinical specimens. J. Clin. Microbiol., 27: 1538-1542.
PubMedDirect Link - Welch, D.F., 1991. Application of cellular fatty acid analysis. Clin. Microbiol. Rev., 4: 422-438.
PubMedDirect Link - Woese, C.R., E. Stackebrandt, T.J. Macke and G.E. Fox, 1985. A phylogenetic definition of the major eubacterial taxa. Syst. Appl. Microbiol., 6: 143-151.
PubMed - Duremdez, R., A. Al-Marzouk, J.A. Qasem, A. Al-Harbi and H. Gharabally, 2004. Isolation of Streptococcus agalactiae from cultured silver pomfret, Pampus argenteus (Euphrasen), in Kuwait. J. Fish Dis., 27: 307-310.
CrossRefPubMedDirect Link