
M.R. Dayer
Department of Biology, Faculty of Sciences, Shahid Chamran University of Ahvaz, Ahwaz, Iran
I. Safari
Department of Biology, Faculty of Sciences, Shahid Chamran University of Ahvaz, Ahwaz, Iran
M.S. Dayer
Department of Parasitology and Medical Entomology,Tarbiat Modares University, Tehran, Iran
Pakistan Journal of Biological Sciences
Year: 2009 | Volume: 12 | Issue: 14 | Page No.: 1025-1030







ABSTRACT
In the present study, the effects of administrating 4 mM and 300 mg kg-1 b.wt. of quinolinic acid were studied, in vitro and in vivo, respectively, to evaluate its inhibitory activity on phosphoenolpyruvate carboxykinase in diabetic rats. The results of in vitro studies have clearly indicated the inhibitory effect of quinolinic acid on enzyme activity. The hill plot showed the binding stoichiometry of quinolinic acid per enzyme to be 4:1. The in vivo studies showed that intra peritoneal injection of 300 mg kg-1 b.wt. initiates reduction of blood glucose level in 1 h after injection, restoring the blood glucose to its normal level at 2 h post injection and keeping it constant for at least further 4 h. Based on present results we concluded that quinolinic acid and hence its precursor tryptophan having induced obvious hypoglycemic effects in normal and diabetic rats at high enough concentrations, they are worthy of further study and research for their hyperglycemic effect in other diabetic animal models.
PDF Abstract XML References Citation
How to cite this article
M.R. Dayer, I. Safari and M.S. Dayer, 2009. New Evidence on Hypoglycemic Effect of Quinolinic Acid in Diabetic Rats. Pakistan Journal of Biological Sciences, 12: 1025-1030.
DOI: 10.3923/pjbs.2009.1025.1030
URL: https://scialert.net/abstract/?doi=pjbs.2009.1025.1030
DOI: 10.3923/pjbs.2009.1025.1030
URL: https://scialert.net/abstract/?doi=pjbs.2009.1025.1030
INTRODUCTION
Phosphoenolpyruvate carboxykinase, PEP- carboxykinase EC number 4.1.4.32, catalyzes the first committed rate limiting step in gluconeogenesis (Nordlie and Lardy, 1963). This reaction involves carboxylation of oxalacetate to phosphoenolpyruvate (PEP) using one nucleoside triphosphate (GTP or ITP) and two equivalent of cations such as Mn2+ or Fe2+:
The reaction is an important step in the formation of glucose from non carbohydrate carbon sources via gluconeogenesis. Cellular localization of PEP- carboxykinase is species dependent, however, it is predominantly found in the cytoplasmic fraction of liver cells in rat, mouse and hamster (Nordlie and Lardy, 1963; Ballard and Honson, 1967; Soling and Kleineke, 1976), in the mitochondrial fraction of chicken and pigeon cells and in both fractions of liver cells in rabbit, pig and human (Nordlie and Lardy, 1963; Holten and Nordlie, 1965). The PEP-carboxykinase plays its physiological role through participation in glucose production in fasting conditions (Landau et al., 1996; Katz and Tayek, 1998; Granner and Pilkis, 1990; Jungermann,1992; Jungermann and Kietzmann, 1996). While, cytoplasmic isoform of PEP-carboxykinase responds to hormonal control, the mitochondrial isoform remains unchanged under different physiological conditions (Xu et al., 2005; Hanson and Reshef, 1997; Drewnowsk et al., 2002). The over expression of cytoplasmic isoform under diabetic or fasting states and the blockage of its biosynthesis in feeding states by insulin induced mechanism have been extensively reviewed (Rajas et al., 2000; Beale et al., 2002; Franckhauser et al., 2002). The increased activity of PEP-carboxykinase in diabetes causes hyperglycemia in both types of diabetes namely IDDM (Insulin Dependent Diabetes Mellitus) and NIDDM (Non Insulin Dependent Diabetes Mellitus) (DeFronzo and Ferrannini, 1991; Consoli et al., 1989; Beale et al., 2002). Nevertheless, the cytoplasmic isoform of PEP-carboxykinase is necessary for life so that its complete absence results in persistent hypoglycemia and finally in death (Hakimi et al., 2005; She et al., 2000, 2003; Vidnes and Sovik, 1976; Hanson and Reshef, 1997; Beale et al., 2002). It has been shown that inhibition of PEP-carboxykinase decreases diabetic hyperglycemia suggesting that its inhibitors can be of pharmacological potential in treating the diabetic hyperglycemia. Inhibition through post transcriptional gene silencing by vector RNAi was reported to cause a reduction in glucose production (Gómez-Valadés et al., 2006). On the other hand, tryptophan administration to fasted and diabetic rat resulted in hypoglycemic effect.
The inhibitory effects of tryptophan on gluconeogenesishas been attributed to quinolinic acid, the well known metabolite of tryptophan, which requires 40 min lag time for its metabolic production (Snoke et al., 1971). However, there exist conflicting reports about the inhibitory effects of tryptophan and quinolinic effects on PEPCK enzyme and the resulting hypoglycemic effect (Alvares and Ray, 1974). In the present study we comparatively studied the previous reports on quinolinic acid and examined its inhibitory potency in vivo and in vitro on gluconeogenesis in diabetic rat.
MATERIALS AND METHODS
This study is the result of research project conducted from 12 March 2008 to 10 Feb 2009 in the Department of Biology of Shahid Chamran University of Ahvaz.
All chemicals were purchased from sigma chemical Co. and from Aldrich chemical Co.
Diabetes production: Diabetes was produced in rat by intra peritoneal injection of 100 mg kg-1 of body weight Streptozotocin (STZ) dissolved in 0.9% sodium chloride. Diabetic rats with blood glucose values at over 300 mg/100 mL, determined by glucose oxidase method, were used as a diabetic model one week after the STZ injection.
Injection of inhibitor: Quinolinic acid at 300 mg kg-1 b.wt. dose were suspended in an appropriate volume of 0.9% sodium chloride and injected intraperitoneally to the animals. The quinolinic acid was injected to seven diabetic rats and the same volume of saline was also injected to 5 diabetic rats as control group. The blood glucose changes were monitored using glucose oxidase method to measure blood glucose level as descried in glucose determination part. The same protocol were used to show the decreased activity of PEP-carboxykinase in the presence of inhibitors, in this case animals were killed 2 h after the injection time with a blow to the head, their livers quickly removed and placed in ice-cold 0.25 M sucrose for enzyme purification.
Glucose determination: Blood glucose was determined using oxidase/peroxidase (GOD-POD) method as described by Trinder (1969).
Enzyme purification: PEP-carboxykinase was purified from rat liver cytosol according to the method of Balard and Hanson (1969) and Chang and Lane (1966) as follows: twenty Wistar rats weighting about 300 g were starved for 24 h prior to killing. This period of starvation increases the amount of PEP-carboxykinase activity. All the rat's livers were homogenized in 2 volume of 0.25 M sucrose, containing 0.05 M Tris-HCl buffer (pH 7.4), with a coaxial homogenizer and centrifuged at 100,000 g for 1 h to obtain 450 mL of clear supernatant. To obtain ammonium sulfate fractionation of enzyme, sufficient solid ammonium sulfate was added to the supernatant from the previous step to bring the solution to 45% saturation. It was usually necessary to add some 1 M Tris during this fractionation to maintain the pH of enzyme solution at 7.0. After standing for 15 min the suspension was centrifuged at 5000 g for 15 min and the precipitate was discarded. Additional solid ammonium sulfate was added to increase the percentage saturation of the solution to 65% and after standing for 15 min this suspension was centrifuged as before. The precipitate was dissolved in 0.05 M Tris-chlorid, pH 8 to give a volume of approximately 75 mL. All steps in the purification were carried out at 0-4°C to avoid instability of the enzyme at higher temperatures.
Enzyme assay: The assay used in all experiments was essentially as reported by Chang and Lane and modified by Ballard and Hanson (1969) and Chang and Lane (1966). The enzyme activity was determined in conversion of oxalacetate to phosphoenolpyruvate direction. This reaction is coupled with malate dehydrogenase reaction i.e., excess amount of malate, NAD+ and malate dehydrogenase were taken in the reaction mixture to provide enough amount of oxalacetate as enzyme substrate for enzyme assay. The 2 mL reaction mixture contained 240 units of malate dehydrogenase, 2 mM MnCl2, 0.0536 g malate, 100 μL of purified PEP-carboxykinase and 0.0527 g NAD+ were incubated in 200 mM Tris-HCl pH7.5 in a water bath 37°C for 1.5 h. After this period of time the absorbance change at 340 nm was constantly zero. Then the buffer pH was brought to 8 by adding enough sodium hydroxide (optimum pH of PEP-carboxykinase). The oxalacetate formed from malate dehydrogenase was determined spectrophotometrically by measuring the NADH generated in the reaction after equilibration at 37°C for 4 min. Nine micro of GTP was added to start the reaction. Initial rates were taken from the linear portion of the recording to measure the enzyme activity. The specific activity of PEP-carboxykinase is expressed in nmol phosphoenolpyruvate formed per min per mg protein. Enzyme assay was carried out using a Shimadzu model UV-3100 (Japan) spectrophotometer and a thermostatically controlled cell compartment with a Haak D8 water bath.
Statistical analysis: The data expressed as Mean±SEM, the statistical analysis was performed using one way Analysis of Variance (ANOVA). The level of statistical significance was set at p<0.05. Calculations were performed using the SPSS statistical package.
RESULTS AND DISCUSSION
In an in vitro inhibitory experiment the effects of 4 mM concentrations of quinolinic acid were examined in optimum conditions on purified PEP-carboxykinase activity. In Fig. 1 the inhibitory effect of quinolinic acid on PEP-carboxykinase has been presented as enzyme activity. The remaining activity of enzyme in the presence of Quinolinic acid was 18.4 nmol min-1 mg which was equal to 63% inhibition in enzyme activity.
In vitro effect of different concentrations of quinolinic acid on enzyme activity is depicted in Fig. 2.
It is obvious that at 4 mM concentration, the enzyme activity was completely inhibited. The Hill plots of enzyme activity in the presence of quinolinic acid shows four binding sites for quinolinic acid on PEP-carboxykinase. Also, the inhibitory constant of quinolinic acid (Ki) was shown to be equal to as previously reported by Snoke et al. (1971) and McDaniel et al. (1972).
Figure 3 shows the effect of quinolinic acid injected intraperitoneally at 300 mg kg-1 b.wt. to diabetic and normal rats a week after induction of diabetes, depicting blood glucose level and its changing trend in time.
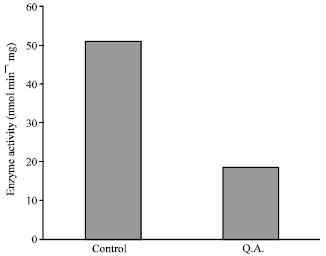 | |
| Fig. 1: | Enzyme activity of purified PEP-carboxykinase in the presence and absence of 4 mM concentration of quinolinic acid in Tris buffer, pH: 8, 200 mM concentration, 37°C temperature and 4 min incubation time |
The inhibitory effect of quinolinic acid began at 1.5 h post-injection causing blood glucose decline with time, which continued for 3 more hs before its recovery (data not shown) probably because of degradation of quinolinic acid by rat liver. The most evident hypoglycemic effect on blood glucose was observed about 3 h after injection.
As shown in Fig. 4 intraperitoneal injection of quinolinic acid at 300 mg kg-1 b.wt. dose cause a significant reduction in PEP-carboxykinase activity.
Gluconeogenesis is a biochemical pathway by which glucose is synthesized in fasting conditions from non carbohydrate precursors.
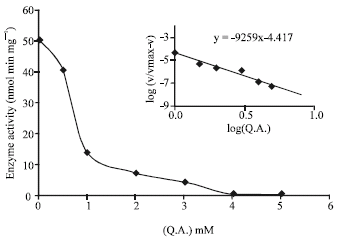 | |
| Fig. 2: | Effect of different concentrations of quinolinic acid on PEP-carboxykinase activity. Tris buffer, pH: 8, 200 mM concentration, 37°C temperature and 4 min incubation Time. Hill plot is shown inside the curve |
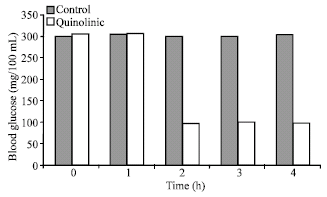 | |
| Fig. 3: | Alteration of glucose homeostasis in diabetic and control rat following intraperitoneal injection of 300 mg kg-1 of body weight of quinolinic acid to diabetic 24 h fasted rats. The blood glucose determined by enzymatic method in samples obtained from the tail vein. The values are expressed as the Mean±SE, p<0.05 for seven animals in each groups |
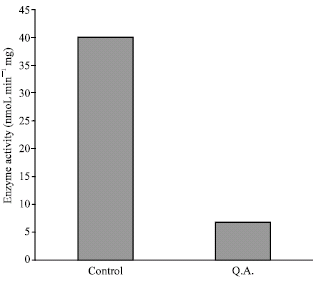 | |
| Fig. 4: | In vivo determination of PEP-carboxykinase activity after injection of 300 mg kg-1 of body weight quinolinic acid. Seven diabetic rats in treated group and five diabetic rats in control group were chosen. Animals killed 2 h after injection and the PEP-carboxykinase extracted and its activity determined as described in methods. The values are expressed as the Mean±SE, p<0.05 |
Three key enzymes namely PEP-carboxykinase, Fructose-1,6-bisphosphatase (FBPase) and glucose-6-phosphatase are involved in this pathway (Barthel and Schmoll, 2003) the former of which, PEP-carboxykinase, catalyzes the first rate limiting step of gluconeogenesis. In fed state, insulin is the most important factor inhibiting gluconeogenesis via suppressing biosynthesis of glucogenic enzymes. In low carbohydrates diet, fasting or diabetes, glucogenic enzymes increase dramatically. The failure in insulin biosynthesis in diabetes results in over production of glucogenic enzymes, accelerating gluconeogenesis and consequently hyperglycemia. Thus, the inhibition of gluconeogenesis, e.g., by enzyme inhibitors in diabetes is of great interest, because of its compensatory effect on hyperglycemia. The PEP-carboxykinase, in contrast to other glucogenic enzymes, is placed in a much more strategic point allowing a metabolic sink of non carbohydrates intermediates to inter the gluconeogenic pathway. This is why PEP-carboxykinase inhibition has been subject of numerous studies for drug design purposes. Tryptophan and its metabolites have vastly been studied for their hypoglycemic effects. Smith and Pogson (1977) have shown that injection of tryptophan decreases glucose production in fed and fasted rats. They observed a 40 min lag time in tryptophan hypoglycemic effect, which was proposed to be the time required for quinolinic acid production via kynurenine pathway (Bender, 1985). Quinolinic acid inhibits PEP-carboxykinase by cooperative manner with Ki values ranging from 0.039 to 0.078 mM (McDaniel et al., 1972). The results also shown that the hypoglycemic effect of quinolinic acid ceases after 1.5 h because of its metabolic degradation. Elliot et al. (1977) studied the uptake of quinolinic acid by rat liver. They showed that the rate at which quinolinic acid enters rat liver cells was low, measuring about 0.0136 mM min-1. However, (Alvares and Ray, 1974) studies came up with three important findings. First, diabeticconditions increased phosphoenolpyruvate carboxykinase activity so that higher concentrations of inhibitors were necessary to produce the same inhibitory effects. Smith et al. (1978) obtained inhibitory response at higher concentrations of inhibitor. Cook and Pogson (1983) also reported that oxaloacetate, the substrate of PEP-carboxykinase, increases more in diabetic than in fasting conditions. Assuming the cooperative mechanism of quinolinic acid inhibition, the presence of increased concentrations of oxaloacetate prevents quinolinic acid action. Second, there was a significant time course inactivation of quinolinic acid, where pre incubation of perfused liver with quinolinic acid showed the least decrease in glucose production from lactate. However the simultaneous addition of quinolinic acid and lactate showed more inhibition in glucose production. Considering these findings, the contradictions about quinolinic acid effects on gluconeogenesis could be understood. To shed light on this ambiguity, we decided to re-examine the inhibitory effect of relatively higher dose of quinolinic acid using in vivo trials with diabetic animals. Our in vitro experiments (Fig. 1) showed that Quinolinic acid at 4 mM concentration was required for inducing significant inhibition of PEP-carboxykinase. The Hill plot shows that binding stoichiometry of Quinolinicacid/PEP-carboxykinase is 4:1 and the:
It is clear that due to competitive behavior of quinolinic acid, ensuring its effective concentrations is a determinant parameter in achieving the expected results. From careful study of procedures used in others works, wecame up with the hypothesis that increased gluconeogenesisflux, increased xaloacetateconcentrations and therefore increased PEP-carboxykinase activity may require a higher concentration of quinolinic acid, if hypoglycemic effect in diabetic animals was to be achieved. This was found to be at more than 200 mg kg-1 b.wt. of quinolinic acid. As depicted in Fig. 2, the blood glucose began to decrease after 1 h reaching the normal
level after 2 h and remains constant for at least 4 h before resuming to increase to higher levels. In this work, we decided to check whether the PEP-carboxykinase activity is changed after I.P. injection of 300 mg kg-1 b.wt. or not. In other word, we wanted to know if the quinolinic acid acts via PEP-carboxykinase inhibition or other mechanism. Figure 4 shows the result of this experiment. PEP-carboxykinase activity is diminished from 40 nmol min-1 mg to about 6.7 nmol min-1 mg after 2 h. Present data clearly suggest that quinolinic acid or its precursor tryptophan has a prominent hypoglycemic effects in decreasing glucose level in diabetic rats and causing a significant relief in diabetic hyperglycemia and could be considered as a hypoglycemic drug for more precise experimental and clinical trails and we suggest it as a food additive (especially tryptophan amino acid) for preventing hyperglycemic conditions and diabetic consequences.
ACKNOWLEDGMENT
The financial support of Chamran University of Ahwas is acknowledged.
REFERENCES
- Alvares, F.L. and P. Ray, 1974. Lack of inhibition by L-tryptophan or quinolinate of gluconeogenesis in diabetic rats. J. Biol. Chem., 249: 2058-2062.
Direct Link - Ballard, F.J. and R.W. Honson, 1967. Phosphoenolpyruvate carboxykinase and pyruvate carboxylase in developing rat liver. Biochemistry, 104: 866-871.
Direct Link - Ballard, F.J. and R.W. Hanson, 1969. Purification of phosphoenolpyruvate carboxykinase from the cytosol fraction of rat liver and the immunochemical demonstration of differences between this enzyme and the mitochondrial phosphoenolpyruvate carboxykinase. J. Biol. Biochem., 244: 5625-5630.
Direct Link - Barthel, A. and D. Schmoll, 2003. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab., 285: E685-E692.
Direct Link - Beale, E.G., R.E. Hammer, B. Antoine and C. Forest, 2002. Glyceroneogenesis comes of age. Faseb J., 16: 1695-1696.
Direct Link - Chang, H.C. and M.D. Lane, 1966. The enzymatic carboxylation of phosphoenol-pyruvate. ІІ. Purification and properties of liver mitochondrial phosphoenolpyruvate carboxycinase. J. Biol. Chem., 241: 2413-2420.
Direct Link - Consoli, A., N. Nurjhan, F. Capani and J. Gerich, 1989. Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes, 38: 550-557.
Direct Link - Cook, J.S. and C.I. Pogson, 1983. Tryptophan and glucose metabolism in rat liver cells. The effect of DL-6-chlorotryptophan, 4-chloro-3-hydroxyanthranilate and pyrazinamide. Biochem. J., 214: 511-516.
Direct Link - DeFronzo, R.A. and E. Ferrannini, 1991. Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia and atherosclerotic cardiovascular disease. Diabetes Care, 14: 173-194.
PubMedDirect Link - Drewnowsk, K.D., M.R. Craig, S.R. Digiovanni, J.M. McCarty, A.F. Moorman, W.H. Lamers and A.C. Schoolwerth, 2002. PEPCK mRNA localization in proximal tubule and gene regulation during metabolic acidosis. J. Physiol. Pharmacol., 53: 3-20.
PubMed - Franckhauser, S., S. Munoz, A. Pujol, A. Casellas and E. Riu et al., 2002. Increased fatty acid re-esterification by PEPCK overexpression in adipose tissue leads to obesity without insulin resistance. Diabetes, 51: 624-630.
Direct Link - G�mez-Valades, A.G., A. Vidal-Alabr�, M. Molas, J. Boada, J. Berm�dez, R. Bartrons and J.C. Perales, 2006. Overcoming diabetes-induced hyperglycemia through inhibition of hepatic phosphoenolpyruvate carboxykinase (GTP) with RNAi. Mol. Therapy, 13: 401-410.
Direct Link - Granner, D. and S. Pilkis, 1990. The genes of hepatic glucose metabolism. J. Biol. Chem., 265: 10173-10176.
Direct Link - Hakimi, P., M.T. Johnson, J. Yang, D.F. Lepage and R.A. Conlon et al., 2005. Phosphoenolpyruvate carboxykinase and the critical role of cataplerosis in the control of hepatic metabolism. Nutr. Metabol., 2: 33-44.
Direct Link - Hanson, R.W. and L. Reshef, 1997. Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu. Rev. Biochem., 66: 581-611.
CrossRef - Holten, D.D. and R.C. Nordlie, 1965. Comparative studies of catalytic properties of guinea pig liver intra-and extramitochondrial phosphoenolpyruvate carboxykinases. Biochemistry, 4: 723-731.
Direct Link - Jungermann, K., 1992. Role of intralobular compartmentation in hepatic metabolism. Diabete Metab., 18: 81-86.
PubMed - Jungermann, K. and T. Kietzmann, 1996. Zonation of parenchymal and non-parenchymal metabolism in liver. Annu. Rev. Nutr., 16: 179-203.
Direct Link - Katz, J. and J.A. Tayek, 1998. Gluconeogenesis and the Cori cycle in 12-,20-and 40-h fasted humans. Am. J. Physiol., 275: E537-E542.
Direct Link - Landau, B.R., J. Wahren, V. Chandramouli, W.C. Schumann, K. Ekberg and S.C. Kalhan, 1996. Contributions of gluconeogenesis to glucose production in the fasted state. J. Clin. Invest., 98: 378-385.
Direct Link - Nordlie, R.C. and H.A. Lardy, 1963. Mammalian liver phosphoenolpyruvate carboxykinase activities. J. Biol. Chem., 238: 2259-2263.
Direct Link - Rajas, F., M. Croset, C. Zitoun, S. Montano and G. Mithieux, 2000. Induction of PEPCK gene expression in insulinopenia in rat small intestine. Diabetes, 49: 1165-1168.
Direct Link - She, P., M. Shiota, K.D. Sholton, R. Chalkley, C. Postic M.A. Magnuson, 2000. Phosphoenolpyruvate carboxykinase is necessary for the integration of hepatic energy metabolism. Mol. Cell. Biol., 20: 6508-6517.
Direct Link - She, P., S.C. Burgess, M. Shiota, P. Flakoll and E.P. Donahue et al., 2003. Mechanisms by which liver-specific PEPCK knockout mice preserve euglycemia during starvation. Diabetes, 52: 1649-1654.
Direct Link - Smith, S.A. and C.I. Pogson, 1977. Tryptophan and control of plasms glucose concentrations in the rat. Biochem. J., 168: 495-506.
Direct Link - Smith, S.A., K.R. Elliott and C.I. Pogson, 1978. Differential effects of tryptophan on glucose synthesis in rats and guinea pigs. Biochem J., 176: 817-825.
Direct Link - Vidnes, J. and O.Sovik, 1976. Gluconeogenesis in infancy and childhood. Acta Pediatr. Scand., 65: 307-312.
PubMed - Xu, H., Q. Yang, M. Shen, X. Huang and M. Dembski et al., 2005. Dual specificity MAPK phosphatase 3 activates PEPCK gene transcription and increases gluconeogenesis in rat hepatoma cells. J. Biol Chem., 280: 36013-36018.
Direct Link - Trinder, P., 1969. Determination of glucose in blood using glucose oxidase with an alternative oxygen acceptor. Ann. Clin. Biochem., 6: 24-27.
CrossRefDirect Link - Elliott, K.R., C.I. Pogson and S.A. Smith, 1977. Permeability of the liver cell membrane to quinolinate. Biochem. J., 164: 283-286.
PubMedDirect Link - McDaniel, H.G., W.J. Reddy and B.R. Boshell, 1972. The mechanism of inhibition of phosphoenolpyruvate carboxylase by quinolinic acid. Biochim. Biophys. Acta, 276: 543-550.
PubMed - Snoke, R.E., J.B. Johnston and H.A. Lardy, 1971. Response of phosphopyruvate carboxylase to tryptophan metabolites and metal ions. Eur. J. Biochem., 24: 342-346.
CrossRefDirect Link