
H. Momtaz
Department of Microbiology, Faculty of Veterinary Medicine, Islamic Azad University of Shahrekord, Shahrekord, Iran
F. Hemmatzadeh
Department of Microbiology, Faculty of Veterinary Medicine, University of Tehran, P.O. Box 1166, Tehran, Iran
H. Keyvanfar
Department of Microbiology, Faculty of Veterinary Medicine, University of Tehran, P.O. Box 1166, Tehran, Iran
Pakistan Journal of Biological Sciences
Year: 2008 | Volume: 11 | Issue: 20 | Page No.: 2433-2437







ABSTRACT
Bovine leukemia virus (BLV) is a member of the family Retroviridae, genus Deltaretrovirus that has three important gene including gag, pol and env. This virus causes B-cell lymphocytosis and lymphosarcoma in cows. In the first step PCR product of gag gene of BLV isolated in different regions of Iran and BLV-FLK strain were cloned in to a pTZ57R/T vector, then insert were digested by BglII and XhoI restriction enzymes and cloned in to pET-28(a) as an expression vector. For the expression of p24 protein, the pET-28(a) recombinant vector was transformed in BL21(DE3) strain of E. coli competent cell and after induction of the protein having been expressed by IPTG, the presence of gag expressed protein was shown in immunoblotting and SDS-PAGE system. With respect to the remarkable frequency of infection to BLV in Iran and the necessity of controlling it through vaccination with recombinant vaccines of gp51, manufacturing and applying the recombinant p24 protein are vital goals in recognition and distinction between infection and responses caused by vaccine.
PDF Abstract XML References Citation
How to cite this article
H. Momtaz, F. Hemmatzadeh and H. Keyvanfar, 2008. Expression of Bovine leukemia virus p24 Protein in Bacterial
Cell. Pakistan Journal of Biological Sciences, 11: 2433-2437.
DOI: 10.3923/pjbs.2008.2433.2437
URL: https://scialert.net/abstract/?doi=pjbs.2008.2433.2437
DOI: 10.3923/pjbs.2008.2433.2437
URL: https://scialert.net/abstract/?doi=pjbs.2008.2433.2437
INTRODUCTION
Enzootic bovine leukosis (EBL) is an infectious lymphoproliferative disease of cattle, caused by the retrovirus Bovine leukemia virus (BLV) (Bicka et al., 2001).
Like other complex reteroviruses the BLV genome contains the gag, pol and env structural genes and regulatory genes (Sagata et al., 1985; Bicka et al., 2001; De Giusppe et al., 2004; Van den Heuvel et al., 2003, 2005). Most of the structural protein of BLV are immunogenic but the naturally infected animals develop antibodies to env encoded glycoproteins gp51 and gp30 as well as to gag encoded proteins p 24 and p15 (Bicka et al., 2001; Deshayes et al., 1980).
Since the presence of antibodies to BLV is a constant and early feature of BLV infection, serological examination of cattle sera is the best method for detection of infected animals. Most commonly used serological tests are the Agar Gel Immuno Diffusion (AGID) and the Enzyme Linked Immuno Sorbent Assay (ELISA). However, variable results have been obtained by the two methods using the p24 and gp51 antigen. The disparities were often the result of differences in specificity and sensitivity of the tests used. AGID is less sensitive and is not useful for detection of antibodies to p24 antigen. ELISA has been shown to detect both anti-gp51 and anti-p24 antibody with equal sensitivity but this method is prone to generate nonspecific reactions. While in the ELISA the non specific reactions are difficult to distinguish from specific ones, western blot analysis allows precise resolution of the two reactions. So far limited studies have been performed to confirm the usefulness of the immunoblotting assay in the routine serological detection of BLV antibodies (Bicka et al., 2001; Kittelberger et al., 1999; Simard et al., 2000). Recently, the recombinant viral proteins have been found to be widely applicable in immunoassays for detection of specific antibodies. In particular, the use of the recombinant proteins synthesized in E. coli has been well documented in retroviral serology (Bicka et al., 2001; De Giusppe et al., 2004; Van den Heuvel et al., 2003, 2005).
In this study, the gag gene of BLV which encodes protein p24 from Iranian isolated virus was cloned and expressed as 6xHis-p24 fusion protein in E. coli. The preparing recombinant protein will be applied in near future for designing Dot-ELISA kit for detection of antibodies against p24 antigen of Bovine leukemia virus in infected and vaccinated cows.
MATERIALS AND METHODS
This study was conducted from April 2007 to February 2008 in Faculty of Veterinary Medicine, Islamic Azad University of Shahrekord.
Sample, plasmids and bacterial strains: The extracted DNA from buffy coat of one of the BLV infected cows which had previously shown positive molecular and serological results based on ELISA and PCR was selected to be cloned (Momtaz and Hemmatzadeh, 2003). Plasmid pTZ57R/T (Ins T/A clone PCR Cloning kit, Fermentas) and E. coli strain JM107 (Fermentas) were used for initial cloning, sequencing and maintenance of DNA fragment. For recombinant protein production, a prokaryotic expression vector pET-28(a) (Novagen) was used. This vector can express a fusion protein with a six histidin tag (6xHis), a thrombin recognition site and a T7 tag at the N-terminus. The recombinant pET-28(a) (pET-28-gag) is transformed into E. coli BL21 (DE3) (Fermentas) as host strain. The required antibiotics were added to LB media according to the reference recommendation (Sambrook et al.,2001).
Primers design: Perimers were designed according to the published sequence for gag gene of BLV (accession No.: M10987) (Rice et al., 1985). The forward primer, gag F: 5-GGC AGA TCT TGG GAA ATT CCC CCT CCT ATA-3 contain Bgl�? site. Reverse primer, gag R:5-CCG CTG GAG TAG TTT TTT GAT TTG AGG GTT GG-3 contain recognition site for XhoI . The restriction enzyme sites (underlined) were added to the primers for subsequent cloning procedure.
Gene amplification of gag ( encoding the p24 protein): PCR was performed in a 50 μL total volume containing 1 μg of template DNA, 1.5 μM of each primer, 1.5 mM MgCl2 , 150 μM dNTP, 1x PCR buffer and 1.5 unit of Taq DNA polymerase (Sigma). The following conditions were used for amplification: initial denaturation at 95°C for 4 min, followed by 30 cycles of denaturation at 94°C for 1 min, annealing at 56°C for 1 min and extension at 72°C for 50 sec. The program followed by a final extension at 72°C for 6 min. The PCR product was analyzed by electrophoresis in 1% agarose gel in 1X TBE buffer and visualized by ethidium bromide staining on UV transilluminator. The PCR product was purified by High pure PCR product purification kit (Roche applied science) according to the manufacturer recommendation.
Cloning of gag gene: The PCR product was digested with BglII and XhoI and ligated to pTZ57R/T and pET-28(a), which were digested by the same restriction enzymes, using T4 DNA ligase (Invitrogen) at 14°C over night.
E. coli JM107 and E. coli BL21(DE3) competent cells were prepared by calcium chloride method and were used for transformation of pTZ57R/T-p24 and pET-28(a)-p24 vectors, respectively. The transformed bacteria were selected by screening the colonies on LB media containing antibiotic. The suspected colony was further analyzed by restriction enzyme digestion and PCR (Sambrook et al., 2001).
Expression and purification of recombinant p24 protein: E. coli BL21 (DE3) was transformed with pET-28 (a)-p24 and grown in LB broth supplemented with kanamycin (50 mg mL-1) at 37°C with agitation in order to optimize the expression condition, different concentrations of IPTG (0.5, 0.8, 1 and 1.5 mM) at different bacterial growth rates (OD600 = 0.5, 0.7 and 1) were tested for 3 h and analyzed on 17% SDS-PAGE (Laemmili, 1970). The expressed protein was purified using Ni-NTA column (Qiagene) according to manufacture instructions. Quantity of the purified recombinant p24 protein was analyzed by Bradford methods and subsequently it,s quality was assayed by SDS-PAGE 17% (2.5 μg well-1). In order to analyze the cross-reaction between fused segment of p24 protein with infected sera, an E. coli BL21(DE3) containing pET-28(a) a vector was induced by IPTG
Immunoblot analysis: For western blot analysis, 0.5 μg of purified recombinant p24 protein was used per well. As a negative control, the bacterial lysate from induced E. coli BL21 (DE3) contain pET-28 (a) vector was analyzed by western blot. The gel was blotted on to Polyvinylidine difluoride (PVDF Membrane, Roche Diagnostics GmbH) membrane using transfer buffer containing 25 mM Tris (pH = 8.3), 192 mM glycine and 20% methanol at 55v for 1 h at 4°C. The blotted membrane was blocked with 3% (w/v) BSA in TBST buffer (0.5 M NaCl, 0.02 M Tris pH = 8.5, 0.05% Tween 20) for 1 h at room temperature (RT). Membrane was incubated for 2 h at 37°C with BLV-infected cow serum, diluted 1:25, respectively. Negative serum from disinfected cow was used as control. After reaction the primary antibody, the blotted membranes were washed three times with TBST and incubated with peroxidase conjugated anti-bovine IgG (Sigma) at a 1:2500 dilution in TBST. The blots were then washed three times with TBST and reaction were developed by diamino benzidine (DAB) solution (Sigma).
RESULTS AND DISCUSSION
The proviral DNA of BLV virus from buffy coat of one of the BLV infected cows in Iran was prepared and used as template for amplification and cloning of the gag gene encoding the protein p24. The amplified fragment had the expected size of 1180 bp comparing to 1kb DNA ladder (Fermentas) (Fig. 1).
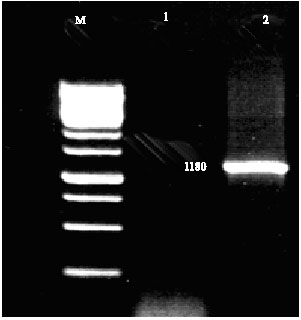 | |
| Fig. 1: | p24 gene amplification by PCR. Lane 1: Molecular weigh marker 1 kb DNA ladder. ; Lane 2: Negative control; Lane 3: Amplified p24 gene |
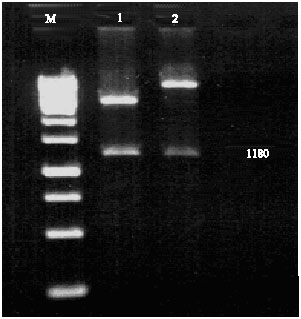 | |
| Fig. 2: | Restriction enzyme analysis of recombinant pTZ57R/T and pET-28(a) plasmids Lane 1: Molecular weight marker 1 kb DNA ladder; Lane 2: Digestion of recombinant pTZ57-R/T plasmid; Lane 3: Digestion of recombinant pET-28(a) plasmid |
The purified PCR product was cloned in pTZ57R/T vector and digestion with BglII and XhoI enzymes. Figure 2 shows recombinant plasmids after digestion.
The recombinant plasmid (pTZ57R/T-p24) was sequenced by specific primers and Sanger sequencing method (Macrogen, Korea). The sequencing result was confirmed by comparing with databases and using basic local alignment search tool (BLAST) software (data not shown).
Expression of pET-28(a)-p24 in E. coli BL21(DE3) induced and the expressed protein was purified by Ni-NTA column (Fig. 3). The result showed that the best conditions for recombinant p24 protein expression can be achieved when 1 mM of IPTG and OD600 = 0.7 for 3 h was used.
To determine the reactivity of recombinant protein p24, the purified recombinant protein was assayed by western blotting method. The five infected cattle serum (which had previously shown positive serological result based on ELISA and AGID) were used. A negative serum from disinfected cattle used as a control. Figure 4 shows the specific interaction between positive sera and purified recombinant p24 protein. There was no reaction between the expressed pET-28(a) in E. coli BL21(DE3) and BLV infected sera (Lane 10 in Fig. 4).
Recognition and study of retrovirus infections are critical in different aspects. The occurrence of mutations consequently genotypic and phenotypic diversity is frequent in retroviruses because of their natural characteristics which is resulted from reverse transcription from their genomes. This feature is that makes the diagnostic value of most of the experimental tests uncertain. There are lots of studies that notice to the special figures and numbers as sensitivity and special quality in diagnostic tests. The repeat of these tests by other researchers usually had different results and it is reported that the reason can be found in genetic diversity of these viruses (Bunqer et al., 1994; Grover and Guillemain, 1992; Bicka et al., 2001).
Anyway, one of the main goals of this examination which was tracing of the coding gene of p24 protein of BLV in the infected samples to this virus, achieved for the first time in Iran and the presence of the corresponded gene was confirmed with the help of sequencing of the fragment.
With respect to this point that primers applied for identification of the gag gene in this study involve the main part of encoding frame of the gene thus, from the beginning the primers were designed for cloning and gene expression of gag in the way that the amplified fragment could be able to be cloned in different vectors such as cloning and expressing vectors.
The second goal of this study was cloning of the mentioned gene in each of the cloning vector (pTZ57R/T vector) and expressing vector (pET-28(a)). The cloning of this gene in the cloning vector after sequencing and comparing resulted sequences to other known sequences of the gag gene available in Genebank indicates the success in cloning the gene into the related vector. Such vector have the capacity to be proliferated in the competent bacterial cells, to be digested because of several sites for restriction enzymes, to be extracted and to be inserted in the expressing vectors. The last finding was derived by cloning the coding gene of p24 protein of BLV in the expressing vector of pET-28(a) for the first time in Iran and the presence of expressing protein was confirmed through SDS-PAGE and immunoblotting system.
 | |
| Fig. 3: | Expression of recombinant p24 protein and its purification Lane 1: Protein marker; Lane 2: pET-28(a)-p24 before induction; Lane 3: pET-28(a)-p24 after induction; Lane 4: Purified p24 recombinant protein |
 | |
| Fig. 4: | Western blot analysis against recombinant p24 protein by BLV-infected sera Line 1: Protein marker; Lane 2: Western blotting pET-28(a)-p24 before induction; Lane 3: Western blotting pET-28(a)-p24 after induction; Lane 4: Western blotting by negative control serum; Lane 5-9: Western blotting by infected sera; Lane 10: Western blotting reaction between the expressed pET-28(a) and positive serum |
Many researchers show that p24 and gp51 proteins which are the products of the gag and env genes of BLV, are strong antigens and usually the first serological responses will be against these antigens. As the gp51 antigen is a very suitable candidate for manufacturing the recombinant vaccines because of its glycoprotein structure and positioning in the membrane of virus. So, Altaner et al. (1991) and Kono et al. (1986 ) have started to design diagnostic methods based on tracing of antibody for p24 antigen and currently several institutions have designed and supplied ELISA and immunoblot methods specific to p24.
With respect to the remarkable frequency of infection to BLV in Iran and the necessity of controlling it through vaccination with recombinant vaccines of gp 51, manufacturing and applying the recombinant p24 protein are vital goals in recognition and distinction between infection and responses caused by vaccine. As the amplified fragment by PCR involves all the domains of p24 and be placed in the expressing frame based on first designs of primers and has successfully been cloned in the expressing vector of pET-28(a) so, the expression of this gene and the preparing recombinant protein will be applied in near future for designing Dot-ELISA kit for detection of antibodies against p24 antigen of bovine leukemia virus in infected and vaccinated cows.
ACKNOWLEDGMENTS
We thank Dr. A. Sharifzadeh, Dr. S. Nekoie, Dr. M. Rohani and Dr. S. Nejat for their cooperation. This study was supported by Grant No. 19594 from the Islamic Azad University of Shahr-e-kord Branch in Iran.
REFERENCES
- Bicka, L., J. Kuzmak, B. Kozaczynska, A. Plucienniczak and A. Skorupska, 2001. Expression of bovine leukemia virus protein p24 in Escherichia coli and its use in the immunoblotting assay. Acta Biochimica Polonica, 48: 227-232.
Direct Link - Bunqer, I., H. Khalaf, C. Cripe and M. Rimpler, 1994. Detection of antibodies against the virus of enzootic bovine leukosis in serum and milk samples using an immunoblot. Dtsch. Tierarztl. Wochenschr., 101: 402-405.
PubMed - Deshayes, L., D. Levy, A.L. Parodi and J.P. Levy, 1980. Spontaneous immune response of bovine leukemia viruse infected cattle against five different viral proteins. Int. J. Cancer, 25: 503-508.
CrossRef - Grover, Y.P. and B. Guillemain, 1992. An immunoblotting procedure for detection of antibodies against bovine leukemia virus in cattle. J. Vet. Med. Ser. B, 39: 48-52.
PubMed - Kittelberger, R., M.P. Reichel, R.M. Meynell, K.M. Tham and J.B. Molloy, 1999. Detection of antibodies against the core protein p24 of the bovine leukaemia virus in cattle for confirmatory serological testing. J. Virol. Methods, 77: 109-114.
Direct Link - Laemmli, U.K., 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227: 680-685.
CrossRefDirect Link - Momtaz, H. and F. Hemmatzadeh, 2003. A serological survey of BLV on cattle in Chaharmahal and Bakhtiary province of Iran. Iran. J. Vet. Res., 4: 37-44.
Direct Link - Rice, N.R., R.M. Stephens, A. Burny and R.V. Gilden, 1985. The gag and pol genes of bovine leukemia virus: Nucleotide sequence and analysis. Virology, 142: 357-377.
CrossRef - Sagata, N., T. Yasuaga, J. Tsuzuku-Kawamura, K. Ohish and Y. Ogawa et al., 1985. Complete nucleotide sequence of the genome of bovine leukemia virus: Its evolutionary relationship to other retroviruses. Proc. Natl. Acad. Sci. USA., 82: 677-681.
CrossRef - Simard, C., S. Richardson, P. Dixon, C. Bélanger and P. Maxwell, 2000. ELISA for the diagnosis of bovine leukosis: Comparision with AGID test approved by the Canadian food inspection agency. Can. J. Vet. Res., 64: 101-106.
Direct Link - Van den Heuvel, M.J., B.J. Jefferson and R.M. Jacobs, 2005. Purified bovine plasma blocking factor decreases bovine leukemia virus p24 expression while increasing protein synthesis and transcriptional activity of peripheral blood mononuclear cells in short-term culture. Can. J. Vet. Res., 69: 186-192.
Direct Link - Van den Heuvel, M.J., D. Portetelle, B. Jefferson and R.M. Jacobs, 2003. Adaptation of a sandwich enzyme linked immunosorbent assay to determine the concentration of bovine leukemia virus p24 and optimal conditions for p24 expression in short-term cultures of peripheral blood mononuclear cells. J. Virol. Methods, 111: 61-67.
Direct Link