
M. Moorthy
Department of Human Anatomy,Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
S. Fakurazi
Department of Human Anatomy,Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
H. Ithnin
Department of Pathology, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
Pakistan Journal of Biological Sciences
Year: 2008 | Volume: 11 | Issue: 15 | Page No.: 1901-1908







ABSTRACT
This study was conducted to identify and to compare the mitochondrial morphological alterations in livers of rats treated with various doses of diclofenac and ibuprofen. Hundred and forty-four male Sprague Dawley rats were dosed with 3, 5 and 10 mg kg-1 diclofenac and ibuprofen in saline via intraperitoneal injection for 15 days. The control group was administered with saline in a similar manner. Four rats were euthanised every 3 days until day 15. While 200 mg kg-1 diclofenac and ibuprofen-treated rats (n = 4) were euthanized 10 h post-treatment. The livers were removed, cleaned and a section across the right lobe was taken and fixed in 4% (v/v) glutaraldehyde for electron microscopy analysis and the remaining samples were kept at -80°C for Western blot analysis. Five milligram per kilogram and 10 mg kg-1 diclofenac-administered rats for 15 days revealed the presence of enlarged mitochondria, irregular and ruptured mitochondrial membranes. While rats administered with 10 mg kg-1 ibuprofen also showed the presence of mitochondria with irregular membrane structure and ruptured membranes. Western blotting analysis of mitochondrial fractions revealed the expression of cytochrome c in all samples and complete absence of cytochrome c expression in the cytosolic fraction of all samples after day 15. Analysis in 200 mg kg-1 diclofenac and ibuprofen-treated groups, revealed expression of cytochrome c in both mitochondrial and cytosolic fractions. This observation indicates that both diclofenac and ibuprofen may alter the morphology of mitochondria, leading to cytochrome c release into the cytosol. Further studies needs to be conducted to investigate on the activity of the mitochondria following both treatments.
PDF Abstract XML References Citation
How to cite this article
M. Moorthy, S. Fakurazi and H. Ithnin, 2008. Morphological Alteration in Mitochondria Following Diclofenac and
Ibuprofen Administration. Pakistan Journal of Biological Sciences, 11: 1901-1908.
DOI: 10.3923/pjbs.2008.1901.1908
URL: https://scialert.net/abstract/?doi=pjbs.2008.1901.1908
DOI: 10.3923/pjbs.2008.1901.1908
URL: https://scialert.net/abstract/?doi=pjbs.2008.1901.1908
INTRODUCTION
Hepatotoxicity is an adverse drug reaction associated with NSAIDs use. Although its occurrence is less common compared to other NSAIDs-related complications, hepatotoxicity is identified as the common cause for withdrawal of some NSAIDs (Teoh and Farrell, 2003).
Diclofenac and ibuprofen are among the most widely used NSAIDs (Al-Nasser, 2000; Boelsterli, 2003). Both have been associated with some form of hepatotoxicity (Bank et al., 1995; Al-Nasser, 2000; Rubeinstein and Laine, 2004; Ponsoda et al., 1995; Masubuchi et al., 2002; Gomez-Lechon et al., 2003a, b).
In the case of diclofenac, significant hepatotoxicity were seen in 1-5 per 100,000 patients consuming this drug (Garcia et al., 1994; Tolman, 1998). Often, the onset of liver injury is characterized by anorexia, nausea, vomiting occurring within 3 months (ranges from 1 to 11 months) (Teoh and Farrell, 2003). Fever and rash have been reported in 25% patients and reaction was severe with jaundice in 50% of the cases (Bank et al., 1995). The hepatotoxicity is believed to be either due to metabolic or immunologic idiosyncrasy (Boelsterli, 2003). 4-OH diclofenac and 5-OH diclofenac yield following metabolism of diclofenac by CYP2C9 and CYP3A4 respectively were identified as toxicants that can undergo covalent binding with various nonprotein or protein groups (Boelsterli, 2003). Many findings also indicate that the metabolites are capable of causing apoptosis of hepatocytes (Ponsoda et al., 1995; Masubuchi et al., 2002; Gomez-Lechon et al., 2003a, b). This was identified to be related to the ability of the drug to cause oxidative stress that is followed by Mitochondrial Permeability Transition (MPT) (Masubuchi et al., 2002). MPT was found to cause leakage of cytochrome c and other apoptotic components from mitochondria into the cytosol, leading to activation of caspase cascade. This event ends with apoptosis of hepatocytes.
Ibuprofen is known to be one of the safest NSAIDs available (Steward, 1992), as not many reports are available on ibuprofen-induced hepatotoxicity. The side effects were identified to be related to the widespread use of the drug (Al-Nasser, 2000). As with other `profen` drugs, acyl glucuronidation is a major route for the biotransformation of ibuprofen (Li et al., 2003) and it is well known that acyl glucuronides are reactive electrophiles that undergo nucleophilic reaction with water, glucuronic acid and glutathione (Grillo and Benet, 2002; Olsen et al., 2002). As with diclofenac, recent study with ibuprofen was also found to induce apoptosis in cell line (Campos et al., 2004). It could be related to the ability of this drug to induce MPT, since it has been shown to target mitochondria (Al-Nasser, 2000). The exact underlying mechanism is still not clear and is under extensive study. This study was conducted to identify and to compare the mitochondrial morphological alterations in the livers of rats treated with various doses of diclofenac and ibuprofen.
MATERIALS AND METHODS
Diclofenac sodium and ibuprofen were purchased from Sigma-Aldrich. Glutaraldehyde 25% EM grade (Agar Scientific Limited, UK), Sodium cocadylate (Agar Scientific Limited, UK), Osmium tetroxide (Agar Scientific Limited, UK). Ethanol absolute (Merck, Germany), 1,2-propyleneoxid (Merck, Germany), Dodecyenyl succinic Anhydride, DDSA (Agar Scientific Limited, UK), Benzyldimethylamine, BDMA (Agar Scientific Limited, UK), Methyl Nadic Anhydride, MDA (Agar Scientific Limited, UK), AGAR 100 Resin (Agar Scientific Limited, UK), Acetone (Ajax Finechem, New Zealand), Lead citrate (Agar Scientific Limited, UK), Uranyl acetate (Agar Scientific Limited, UK), grid 200 mesh (Agar Scientific Limited, UK). Sucrose (Amresco, Ohio), EDTA (Sigma-Aldrich), Glacial acetic acid (ICN Biomedicals, California), Tris-base (Sigma-Aldrich). EDTA (Sigma-Aldrich), BCA protein assay kit (Pierce, USA). Thirty percent Acrylamide solution (Biorad, USA), SDS (Amresco, Ohio), TEMED (Amresco, Ohio), Ammonium Persulfate (Amresco, Ohio), Glycine (Amresco, Ohio), Methanol (Systerm), Glycerol (ICN Biomedical, California), Bromophenol blue (Amresco, Ohio), Dithioreitol, DTT (Amresco, Ohio), Casein (Sigma-Aldrich), NaCl (Merck, Germany), Thimerosal (ICN Biomedicals, California), mouse monoclonal Cytochrome c primary antibody (Santa Cruz Biotechnology Inc., USA), Hydrochloric acid, HCl (ICN Biomedicals, California), rabbit anti-mouse HRP secondary antibody (Zymed, California), Pageruler Prestained Protein ladder (Fermentas, Canada), nitrocellulose membrane (Sigma-Aldrich), Supersignal West Pico Chemiluminescent substrate (Pierce, USA).
Animal study: This study was conducted at Faculty of Medicine and Health Sciences, Universiti Putra Malaysia. A hundred and forty four male Sprague Dawley rats were acclimatised under control condition of humidity with regular light/dark cycle and free access to food and water. The rats were randomly distributed into groups of 3, 5 and 10 mg kg-1 diclofenac and ibuprofen, control group and 4 rats as untreated group. The drugs were dosed intraperitoneally at 0.5 mL rat-1 day-1. The control group was given saline in a similar manner. Four rats from each group were euthanised every 3 days until day 15. While 200 mg kg-1 diclofenac and ibuprofen-treated rats (n = 4) were euthanized 10 h post-treatment. Upon euthanisation, livers were removed, cleaned and weighed. A section across the right lobe was taken and fixed in 4% (v/v) glutaraldehyde, which was processed for transmission electron microscopy analysis. The remaining samples were kept under -80°C for Western blot analysis.
Ultrastructural study: A section across the right lobe was removed, cleaned and weighed in normal saline and sliced into small sections of 1 mm3 thickness. These sections were fixed in 4% (v/v) glutaraldehyde for 24 h then post-fixed in 2% osmium tetroxide. The subsequent process was carried out according to standard procedure for electron microscopy analysis. Infiltration process was carried out with propylene oxide and resin mixture. Then, the samples were embedded in freshly prepared resin mixture. Ultrathin section was prepared and stained with uranyl acetate and lead citrate and viewed using transmission electron microscope to detect ultrastructural changes in mitochondria.
Detection of cytochrome c
Subcellular fractionation: Subcellular fractionation was carried out using cold sucrose buffer (0.25 M sucrose, 15 mM Tris-base, 0.1 mM EDTA, pH adjusted to 6.8 with glacial acetic acid). Liver samples from each dose group were pooled, blotted dry, weighed, minced in 5 volumes of sucrose buffer and homogenised at 1000 rpm (Ultra-Turaxx T8). The homogenate was strained and 3 mL of aliquots were stored at -80°C. The remainder of the homogenate was centrifuged at 600 x gav for 10 min (Eppendorf 5810 R). The supernatant was separated following centrifugation. The post nuclear supernatants were centrifuged at 10,000 x gav for 20 min (Eppendorf 5810 R). The resulting supernatant was separated. The supernatant corresponds to cytosolic fraction (with microsomes), 3 mL aliquots were kept at -80°C. The pellet was washed twice by resuspending in one volume of sucrose buffer and recentrifuging. The resulting pellet that corresponds to mitochondrial fraction was kept at -80°C.
Protein assay: Protein concentrations of the samples were carried out using the BCA protein assay reagent kit (Pierce), according to manufacturer`s instruction.
SDS-PAGE and immunoblotting: Protein samples were solubilised by boiling for 3 min in buffer (125 mM stacking gel buffer, 10% (w/v) SDS, 20% (v/v) glycerol, 0.02% (w/v) bromophenol blue, 0.2 M DTT). SDS-PAGE was carried out using Mini PROTE-N® III Electrophoresis Cell apparatus (Biorad) with 4% (w/v) stacking gel and 10% (w/v) resolving gel using the discontinuous buffer system as described by Laemmili (1970). Protein samples were loaded at 25 μg lane-1 and the gel was run at 100 Volt for approximately 75 min in running buffer (0.025 M Tris, 0.192 M glycine, 0.1% (w/v) SDS, pH 8.3). Following electrophoresis, proteins were electrophoretically transferred to a nitrocellulose membrane in transfer buffer (15.7 mM Tris-base, 120 mM glycine and 20% (v/v) methanol, pH 8.3) at constant voltage of 100 V for 1 h at 4°C using mini trans-blot elctrophoresis cell (Biorad, USA).
Subsequent steps were carried out at room temperature with continuous shaking. Following immunoblotting, the membrane was incubated overnight in blocking buffer (154 mM NaCl, 2.8% (w/v) casein, 10 mM Tris-base and 0.02% (w/v) thimerosal, pH 7.6 with concentrated HCl) to block the non-specific binding sites.
The nitrocellulose membrane was then incubated with mouse monoclonal cytochrome c primary antibody diluted in wash buffer (154 mM NaCl, 0.5% w/v casein, 1 mM Tris, 0.02% w/v thimerosal) (dilution 1:1000). After four 10 min washes to remove unbound antibody, the membrane was then incubated for 2 h with rabbit anti-mouse HRP secondary antibody (1:10,000 in wash buffer). Following series of washes with wash buffer and TBS, the membrane was incubated with Pierce Supersignal West Pico Chemiluminescent substrate reagent for 5 min. The expression of protein bands were captured with Flourochem 5500 (Alpha Innotech).
RESULTS AND DISCUSSION
Ultrastructural changes in mitochondria: Rats treated with 5 and 10 mg kg-1 diclofenac for 15 days showed presence of abnormal mitochondria. The abnormalities observed in 5 mg kg-1 diclofenac administered group include the presence of enlarged mitochondria (Fig. 1), irregular mitochondrial membrane (Fig. 2) and ruptured mitochondrial membranes (Fig. 3). Similarly, 10 mg kg-1 diclofenac given rats revealed presence of enlarged mitochondria (Fig. 4) and ruptured mitochondrial membranes (Fig. 5).
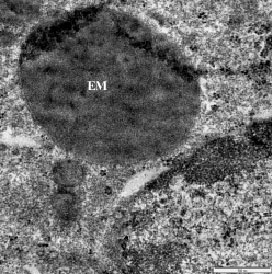 | |
| Fig. 1: | Slight enlargement of mitochondria (EM) in 5 mg kg-1 diclofenac-treated groups after day 15, 8000x |
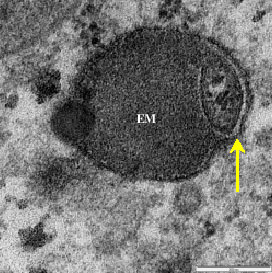 | |
| Fig. 2: | (→) indicates presence of irregular mitochondrial membranes in 5 mg kg-1 diclofenac administered rats after day 15, 20 000 |
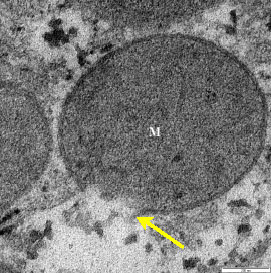
Meanwhile, when rats were administered with ibuprofen, abnormal mitochondrial ultrastructures were seen only with 10 mg kg-1 ibuprofen-dosed group on day 15. The changes observed include, mitochondria with irregular membrane structure (Fig. 6) and ruptured membranes (Fig. 7). Observations conducted on samples of both diclofenac and ibuprofen treated groups at lower doses and at earlier time points showed normal liver ultrastructure similar to saline-treated rats as shown in Fig. 8.
 | |
| Fig. 3: | (→) indicates ruptured mitochondrial membranes in 5 mg g-1 diclofenac-treated rats, 20 000 |
 | |
| Fig. 4: | Slightly enlarged mitochondria (EM) in 10 mg kg-1 diclofenac administered group after day 15, 8000x |
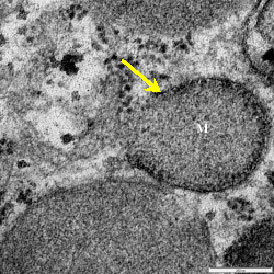 | |
| Fig. 5: | (→) indicates ruptured mitochondrial membrane in 10 mg kg-1 diclofenac injected rat, 20 000 |
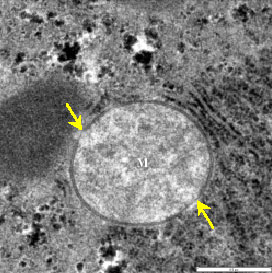 | |
| Fig. 6: | (→) indicates presence of irregular mitochondrial membrane in 10 mg kg-1 ibuprofen administered rats after day 15, 8000x |
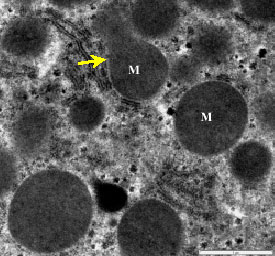 | |
| Fig. 7: | (→) shows ruptured mitochondrial membrane in 10 mg kg-1 ibuprofen injected group, 8000x |
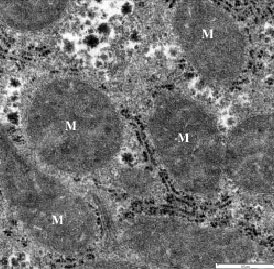 | |
| Fig. 8: | Intact mitochondria (M) in saline-treated rats at day 15, 8000x |
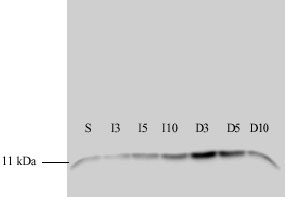 | |
| Fig. 9: | Expression of cytochrome c in mitochondrial fractions of 3-10 mg kg-1 ibuprofen and diclofenac administered rats on day 15 |
 | |
| Fig. 10: | Absence of cytochrome c expression in liver cytosolic fraction of 3-10 mg kg-1 ibuprofen and diclofenac-treated rats on day 15 |
Western blotting analysis to detect cytochrome c expression in liver samples: Western blotting analysis was carried out to detect the possible expression of cytochrome c in the mitochondrial and cytosolic fractions which indicates possible rupture in mitochondrial membranes.
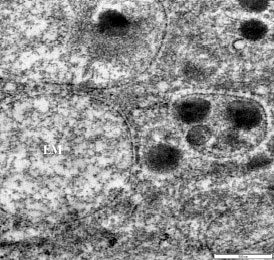
Following subcellular fractionation of the liver homogenates, the expression of cytochrome c was clearly observed in the mitochondrial fraction of diclofenac and ibuprofen-treated groups as shown in Fig. 9. There was no expression of cytochrome c after 15 days of diclofenac and ibuprofen treatment at 3-10 mg kg-1 as indicated in Fig. 10. However, with a single dose of 200 mg kg-1 diclofenac and ibuprofen, cytochrome c is expressed in the cytosolic fraction as revealed in Fig. 11.
Recent studies have identified the ability of diclofenac and ibuprofen to induce apoptosis in various cell lines (Kusuhara et al., 1998; Gomez-Lechon et al., 2003a; Masubuchi et al., 2002; Campos et al., 2004) and mitochondria was found to play a pivotal role in the mechanism (Gomez-Lechon et al., 2003a; Masubuchi et al., 2002; Campos et al., 2004).
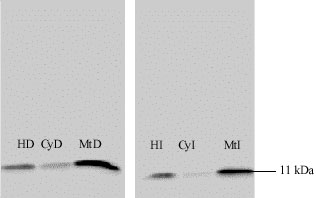 | |
| Fig. 11: | Presence of cytochrome c in liver homogenate (HI), cytosolic fraction (CyI) and mitochondrial fraction (MtI) of 200 mg kg-1 ibuprofen-treated rats and in liver homogenate (HD), cytosolic fraction (CyD) and mitochondrial fraction (MtD) of 200 mg kg-1 diclofenac-treated groups 10 h post-treatment. Saline (S), 3 mg kg-1 ibuprofen (I3), 5 mg kg-1 ibuprofen (I5), 10 mg kg-1 ibuprofen (I10), 3 mg kg-1 diclofenac (D3), 5 mg kg-1 diclofenac (D5), 10 mg kg-1 diclofenac (D10) |
Multiple treatments for 15 days with diclofenac and ibuprofen, induced mitochondrial swelling and mitochondrial membrane rupture. The changes seen in this study suggested to be dose and frequency dependent (Kretz-Rommel and Boelsterli, 1993a, b; Masubuchi et al., 1998). The observation indicates that diclofenac is more potent compared to ibuprofen (Masubuchi et al., 1998; Siraki et al., 2005) in inducing mitochondrial changes. This is in consistent with IC50 of diclofenac for inhibition of COX-1 of 0.26 in comparison to ibuprofen of 5.9 (Cryer and Feldman, 1998).
Mitochondrial swelling and membrane rupture are believed to be due to a phenomenon called Mitochondrial Permeability Transition (MPT). MPT refers to an increase in mitochondrial membrane permeability to solutes with molecular mass less than 1500 Da. The MPT is believed to be formed at the contact sites between the inner and outer mitochondrial membranes. MPT results in depolarization and chemical or solutes equilibration between cytoplasm and mitochondrial matrix. This ultimately leads to disruption and rupture of outer mitochondrial membrane (Benardi et al., 2006, 1999; Crompton, 1999).
Previously, it has been reported that the inability of mitochondria to produce ATP as the major cause of diclofenac-induced hepatotoxicity (Ponsoda et al., 1995; Bort et al., 1999). A recent study also revealed the key role of mitochondrial dysfunction in the pathogenesis of diclofenac-induced hepatic injury due to decrease in ATP caused by MPT (Masubuchi et al., 2002). It has also been proved that inhibition of MPT with specific MPT blockers prevents diclofenac-induced apoptosis both in human and rat hepatocytes (Karpinich et al., 2002; Feldtrauer et al., 2002; Gomez-Lechon et al., 2003a). Diclofenac-activated MPT was identified as the consequence of oxidative damage to pre-existing membrane proteins, since simultaneous incubation of diclofenac-added hepatocytes with antioxidants prevented caspase activation and later apoptosis (Sokol et al., 2001; Vrablic et al., 2001; Masubuchi et al., 2002; Gomez-Lechon et al., 2003a).
Increase in intramitochondrial Ca2+ is also believed to be another factor of MPT. It potentiates binding of cyclophilin D to the matrix side of MPT pore in particular to Adenine Nucleotide Transporter (ANT) (Crompton et al., 1998). Increase in intramitochondrial Ca2+ was related to the presence of diclofenac metabolites (Lim et al., 2006). 4-OH and 5-OH diclofenac can be metabolized to respective reactive intermediates (Shen et al., 1999; Tang et al., 1999a, b; Poon et al., 2001) that generate Reactive Oxygen Species (ROS). ROS is believed to inactivate critical sulfhydryl groups of Ca2+ pumps (Lim et al., 2006), which leads to increase in intramitochondrial Ca2+ concentration.
While for ibuprofen, similar observation to current study was made in isolated mitochondria and was also related to MPT (Al-Nasser, 2000). It was shown that exposure of liver mitochondria to low concentration of ibuprofen resulted in increase swelling and loss of inner mitochondrial membrane potential, which was inhibited by cyclosporine A (CsA); a well-known MPT inhibitor. This indicates the ability of ibuprofen to induce MPT.
Due to detection of mitochondrial swelling and membrane rupture, cytochrome c release from mitochondria into cytosol was assessed since apoptosis was related to presence of cytochrome c in the cytosol (Gomez-Lechon et al., 2003b).
Cytochrome c expression was detected in the mitochondrial fractions of all samples under study. However, there was no detection of cytochrome c expression in the cytosolic fractions of all doses. This is probably due to the cytosolic fractions did not contain any detectable amount of cytochrome c (Herde et al., 2000). This may also be explained by the detection of only few ruptured mitochondria in 5 and 10 mg kg-1 diclofenac and 10 mg kg-1 ibuprofen given groups. This may also be related to the use of monoclonal cytochrome c primary antibody (ab), since this type of antibody is highly specific as compared to polyclonal antibodies. However, the expression of cytochrome c was detected in both mitochondrial and cytosolic fraction when animals were treated with 200 mg kg-1 of diclofenac and ibuprofen. The cytosolic cytochrome c of diclofenac is more densely expressed than that of ibuprofen; indicating diclofenac to be more potent compared to ibuprofen (Kretz-Rommel and Boelsterli, 1993a, b; Masubuchi et al., 1998) in inducing cytochrome c release from mitochondria into cytosol.
Cytochrome c is involved in the formation of membrane potential for the production of ATP (Robertson and Orrenius, 2000). The addition of cytochrome c to cytosolic extract has been shown to be a determining factor in the activation of caspase and later apoptosis (Liu et al., 1996; Zou et al., 1997). Cytochome c release has been identified as the consequence of mitochondrial swelling and membrane rupture (Benardi et al., 1999). This observation indicates that both diclofenac and ibuprofen may alter the morphology of mitochondria, leading to cytochrome c release into the cytosol. Further studies needs to be conducted to investigate on the activity of the mitochondria following both treatments.
ACKNOWLEDGMENTS
The authors would like to acknowledge the Malaysian Toray Science Foundation (MTSF) (Grant No. 54853) and the Ministry of Higher Learning Education of Malaysia for financial assistance (Grant No. 55165) and Unit of Microscopy and Microanalysis, Institute of Bioscience (Universiti Putra Malaysia) for their technical assistance.
REFERENCES
- Al-Nasser, I.A., 2000. Ibuprofen-induced liver mitochondrial permeability transition. Toxicol. Lett., 111: 213-218.
CrossRef - Bank, A.T., H.J. Zimmermann, K.G. Ishak and J.G. Harter, 1995. Diclofenac-associated hepatotoxicity: Analysis of 180 cases reported to the food and drug administration as adverse reactions. Hepatology, 22: 820-827.
CrossRef - Benardi, P., L. Scorrano, R. Colonna, V. Petronilli and F. Di Lisa, 1999. Mitochondria and cell death: Mechanistic aspects and methodological issues. Eur. J. Biochem., 264: 687-701.
PubMed - Benardi, P., A. Krauskopf, E. Basso, V. Petronilli, E. Blalahy-Dysan, F. Di Lisa and M.A. Forte, 2006. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J., 273: 2077-2099.
CrossRef - Boelsterli, U.A., 2003. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity. Toxicol. Applied Pharmacol., 192: 307-322.
CrossRef - Bort, R., K. Mace, A. Boobis, M.J. Gomez-Lechon, A. Pfeifer and J. Castell, 1999. Hepatic metabolism of diclofenac: Role of human CYP in the minor oxidative pathways. Biochem. Pharmacol., 58: 787-796.
CrossRef - Campos, C.B.L., G.R. Degasperi, D.S. Pacifico, L.C. Alberici and R.S. Carreira et al., 2004. Ibuprofen-induced Walker 256 tumor cell death: Cytochrome c release from functional mitochondria and enhancement by calcineurin inhibition. Biochem. Pharmacol., 68: 2197-2206.
CrossRef - Crompton, M., S. Virji and J.M. Ward, 1998. Cyclophilin D binds strongly to complexes of the voltage-dependent anion channels and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem., 258: 729-735.
PubMed - Crompton, M., 1999. The mitochondrial permeability transition pore and its role in cell death. Biochem. J., 341: 233-249.
PubMed - Cryer, B. and M. Feldman, 1998. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med., 104: 413-421.
PubMedDirect Link - Waldmeier, P.C., J.J. Feldtrauer, T. Qian and J.J. Lemasters, 2002. Inhibition of the mitochondrial permeability transition by the non immunosuppressive cyclosporine derivative NIM811. Mol. Pharmacol., 62: 22-29.
CrossRef - Garcia, R.L.A., R. Williams, L.E. Derby, A.D. Dean and H. Jick, 1994. Acute liver injury associated with non-steroidal anti-inflammatory drugs and the role of risk factors. Arch. Int. Med., 154: 311-316.
PubMed - Gómez-Lechón, M.J., X. Ponsoda, E. O’Connor, T. Donato, J.V. Castell and R. Jover, 2003. Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochem. Pharmacol., 66: 2155-2167.
CrossRefDirect Link - Gomez-Lechon, M.J., X. Ponsoda, E. O’Connor, T. Donato, R. Jover and J.V. Castell, 2003. Diclofenac induces apoptosis in hepatocytes. Toxicol. In Vitro, 17: 675-680.
CrossRef - Grillo, M.P. and L.Z. Benet, 2002. Studies on the reactivity of clofibryl-S-acyl-CoA thioester with glutathione in vitro. Drug Metab. Dispos., 30: 55-62.
CrossRefPubMedDirect Link - D'Herde, K., B. DePrest, S. Mussche, P. Schotte, R. Beyaert, R. Van Coster and F. Roels, 2000. Ultrastructural localization of cytochrome c in apoptosis demonstrates mitochondrial heterogeneity. Cell Death Diff., 7: 331-337.
CrossRefPubMedDirect Link - Karpinich, N.O., M. Tafani, R.J. Tothman, M.A. Russo and J.L. Farber, 2002. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J. Biol. Chem., 277: 16547-16552.
CrossRef - Kretz-Rommel, A. and U.A. Boelsterli, 1993. Selective protein adducts to membrane protein in cultured rats hepatocytes exposed to diclofenac: Radiochemical and immunochemical analysis. Mol. Pharmacol., 45: 237-244.
PubMed - Kretz-Rommel, A. and U.A. Boelsterli, 1993. Diclofenac covalent protein binding is dependent on acyl glucuronide formation and is inversely related to P4590-mediated acute cell injury in cultured rat hepatocytes. Toxicol. Applied Pharmacol., 120: 155-161.
CrossRef - Kusuhara, H., H. Matsuyuki, M. Matsuura, T. Imayoshi, T. Okumoto and H. Matsui, 1998. Induction of apoptotic DNA fragmentation by nonsteroidal anti-inflammatory drugs in cultured rat gastric mucosal cells. Eur. J. Pharmacol., 360: 273-280.
CrossRef - Laemmli, U.K., 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227: 680-685.
CrossRefDirect Link - Li, C., M.P. Grillo and L.Z. Benet, 2003. In vivo mechanistic studies on the metabolic activation of 2-phenylpropionic acid in rat. J. Pharmacol. Exp. Thr., 305: 250-256.
CrossRef - Lim, M.S., P.L.K. Lim, R. Gupta and A. Boelsterli, 2006. Critical role of free cytosolic calcium, but not uncoupling, in mitochondrial permeability transition and cell death induced by diclofenac oxidative metabolites in immortalized human hepatocytes. Toxicol. Applied Pharmacol., 217: 322-331.
CrossRef - Liu, X., C.N. Kim, J. Yang, R. Jemmerson and X. Wang, 1996. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell, 86: 147-157.
CrossRefDirect Link - Masubuchi, Y., H. Saito and T. Horie, 1998. Structural requirements for the hepatotoxicity of non-steroidal anti-inflammatory drugs in isolated rat hepatocytes. J. Pharmcol. Exp. Ther., 287: 208-213.
PubMed - Masubuchi, Y., S. Nakayama and T. Horie, 2002. Role of mitochondrial permeability transition in diclofenac-induced hepatocyte injury in rats. Heptology, 35: 544-551.
PubMed - Ponsoda, X., R. Bort, R. Jover, M.J. Gomez-Lechon and J.V. Castell, 1995. Molecular mechanism of diclofenac hepatotxicity: Association of cell injury with oxidative metabolism and decrease in ATP levels. Toxic. In vitro, 9: 439-444.
CrossRef - Poon, G.K., Q. Chen, Y. Teffera, J.S. Ngui and P.R. Griffin et al., 2001. Bioactivation of diclofenac via benzoquinone imine intermediates-identification of urinary mercapturic acid derivatives in rats and humans. Drug Metab. Dispos., 29: 1608-1613.
PubMed - Robertson, J.D. and S. Orrenius, 2000. Molecular mechanism of apoptosis induced by cytotoxic chemicals. Crit. Rev. Toxicol., 30: 609-627.
PubMed - Rubeinstein, J.H. and L. Laine, 2004. Systematic review: The hepatotoxicity of non-steroidal anti-inflammatory drugs. Aliment Pharmacol. Ther., 20: 373-380.
CrossRef - Shen, S., M.R. Marchick, M.R. Davis, G.A. Doss and L.R. Pohl, 1999. Metabolic activation of diclofenac by human cytochrome P450 3A4: Role of 5-hydroxydiclofenac. Chem. Res. Toxicol., 12: 214-222.
PubMed - Siraki, A.G., T. Chevaldina and P.J. O'Brien, 2005. Application of quantitative structure-toxicity relationships for acute NSAID cytotoxicity in rat hepatocytes. Chem. Biol. Inter., 151: 177-191.
PubMedDirect Link - Sokol, R.J., M.S. Straka, R. Dahl, M.W. Devereaux and B. Yerushalmi et al., 2001. Role of oxidant stress in permeability transition induced in hepatic mitochondria by hydrophobic bile acids. Pediatr. Res., 49: 519-531.
PubMed - Adams, S.S., 1992. The propionic acids: A personal perspective. J. Clin. Pharmacol., 32: 317-323.
PubMed - Tang, W., R.A. Stearns, S.M. Bandiera, Y. Zhang and C. Raab et al., 1999. Studies on cytochrome P-450-mediated bioactivation of diclofenac in rats and in human hepatocytes: Identification of glutathione conjugated metabolites. Drug Metab. Dispos., 27: 365-372.
PubMed - Tang, W., R.A. Stearn, R.W. Wang, S.H.L. Chiu and T.A. Baillie, 1999. Roles of human hepatic cytochrome P450s 2C9 and 3A4 in the metabolic activation of diclofenac. Chem. Res. Toxicol., 12: 192-199.
CrossRef - Teoh, N.C. and G.C. Farrell, 2003. Hepatotoxicity associated with non-steroidal anti-inflammatory drugs. Clin. Liver Dis., 7: 401-413.
PubMed - Vrablic, A.S., C.D. Albright, C.N. Craciunescu, R.I. Salganik and S.H. Zeisel, 2001. Altered mitochondrial function and overgeneration of reactive oxygen species precede the induction of apoptosis by 1-O-octadecyl-2-methyl-rac-glycero-3-phosphocoline in p53 defective hepatocytes. FASEB J., 15: 1739-1744.
PubMed - Zou, H., W.J. Henzel, X. Liu, A. Lutschg and X. Wang, 1997. Apaf-1, a human protein homologous to C. Elegens CED-4 participates in cytochrome c dependent activation of caspase 3. Cell, 90: 405-413.
CrossRef