
Habib Onsori
Islamic Azad University, Science and Research Campus, Tehran, Iran
Mohammad Ali Hosseinpour
Department of Biology, Faculty of Sciences, University of Tabriz, Tabriz, Iran
Sheideh Montaser-Kouhsari
Department of Biology, Faculty of Sciences, University of Tehran, Tehran, Iran
Mohammad Asgharzadeh
Hematology and Oncology Research Center, Tabriz University of Medical Sciences, Tabriz, Iran
Abbas Ali Hosseinpour
Tabriz University of Medical Sciences, Tabriz, Iran
Pakistan Journal of Biological Sciences
Year: 2007 | Volume: 10 | Issue: 23 | Page No.: 4299-4302







ABSTRACT
Hemophilia A is an X-linked congenital bleeding disorder caused by factor VIII deficiency. The factor VIII gene is on the long arm of the X chromosome at Xq28 spans 186 kb and consists of 26 exons. In this study to identify defects in the factor VIII gene, Single-Stranded Conformation Polymorphism (SSCP) analysis was used. A novel missense mutation due to T → C transition at codon 153 (TGC) of the factor VIII gene which replace a cysteine with an arginine residue, was found in a patient of North-Western of Iran with sever hemophilia A. Direct sequencing of the amplified fragment was performed to confirm the mutation. This study shows that we can use of Polymerase Chain Reaction (PCR) and silver staining of SSCP methods for detecting most of the point mutations causative hemophilia A.
PDF Abstract XML References Citation
How to cite this article
Habib Onsori, Mohammad Ali Hosseinpour, Sheideh Montaser-Kouhsari, Mohammad Asgharzadeh and Abbas Ali Hosseinpour, 2007. Identification of a Novel Missense Mutation in Exon 4 of the Human Factor VIII Gene Associated with Sever Hemophilia a Patient. Pakistan Journal of Biological Sciences, 10: 4299-4302.
DOI: 10.3923/pjbs.2007.4299.4302
URL: https://scialert.net/abstract/?doi=pjbs.2007.4299.4302
DOI: 10.3923/pjbs.2007.4299.4302
URL: https://scialert.net/abstract/?doi=pjbs.2007.4299.4302
INTRODUCTION
Hemophilia A is an X chromosome-linked recessive bleeding disorder that affects approximately 1 in 10,000 males in all population groups (Miyoko et al., 1991). It is caused by mutations in the factor VIII gene, which is localized on the chromosome Xq28 (Habart, 2005). Factor VIII (FVIII) is a plasma glycoprotein that functions as a cofactor for the serine protease factor IXa in the proteolytic activation of factor X to Xa (Geoffrey et al., 1998; Barry, 2002). FVIII is synthesized as a large single-chain protein with structural domains represented as A1-A2-B-A3-C1-C2. In plasma, FVIII circulates in a noncovalent complex with von Willebrand factor as a heterodimer consisting of a light chain (A3-C1-C2 domains) Me2+-linked to a heavy chain (variable lengths of the B domain and A1-A2 domains). Thrombin converts FVIII to activated FVIII by cleaving at residues 372, 740 and 1689. The activated form of FVIII is markedly unstable, with the decay in activity being due to a dissociation of the A2 subunit from the All A3-C1-C2 dimer. Molecular characterization of hemophilia A is hampered by the large size and complexity of the FVIII gene, which is 186 kb long, has 26 exons and encodes a 9 kb mRNA (Arruda et al., 1995; Dalal et al., 2006). More than 900 different mutations including point mutations, insertions, deletions and inversions have been reported in the factor VIII (MIMno. 306700, HAMSTeRS database http://www.europium.csc.mrc.ac.uk) gene which cause hemophilia A (Berber et al., 2006). Point mutations are the most prevalent type of defect, probably underlying the disease in 90-95% of patients. It occurs in the hemophilias and is comprised of missense mutations, nonsense point mutations and mRNA splice-site point mutations (Bhopale and Nanda, 2003). Thus, to characterize FVIII gene mutations, the complete coding region of the gene has to be analyzed. The situation is further complicated by the fact that one third of the FVIII mutations occur as de novo mutation (Tuddenham et al., 1994). Such diversity makes genetic counseling complex and troublesome (Peake et al., 1993). In this study, with the using of Single-Stranded Conformational Polymorphism (SSCP) and direct sequencing, hemophilia A patients of North-Western of Iran were analyzed for mutations in all 26 exons and introns 1 and 22 of the FVIII gene.
MATERIALS AND METHODS
Samples and DNA extraction: Mutation analysis of the FVIII gene was performed in 35 hemophilia A patients from unrelated families of North-Western of Iran. This research project has been approved by ethical committee. All patients have clinical and biochemical characteristics associated with hemophilia A. Genomic DNA was extracted from 2.5 mL of EDTA anticoagulated peripheral blood by SDS-Proteinase K according to Sambrook et al. (1989).
Amplification of genomic DNA: Polymerase Chain Reaction (PCR) of genomic DNA from patient HA10 and a normal male subject were performed using F8Ex4F: 5'-CATGTTTCTTTGAGTGTACAGTGG-3' as a forward primer and F8Ex4R: 5'-TTCAGGTGAAGGAACACAAATG-3 as a reverse primer. The PCRs were carried out in 25 μL reaction mixture containing 1xPCR buffer, 1.5 mM MgCl2, 0.2 mM dNTP, 10 pmoles of each primer, 0.5 U of Taq DNA polymerase and about 1μg of genomic DNA on a Genius Thermal Cycler. After initial denaturation for 2 min at 95°C, 30 cycles of 30 sec at 94°C, annealing 40 sec at 59°C and extension for 30 sec at 72°C, a final extension for 5 min at 72°C followed. The amplified fragments were detected on 1.5% agarose gel by ethidium bromide staining.
Single-Stranded Conformational Polymorphism (SSCP): After examination by agarose gel electrophoresis, PCR products were analyzed by Single-Stranded Conformational Polymorphism (SSCP) and visualized by silver-staining according to a reported method (Chinchang et al., 2005). After dilution of 1.75 μL of PCR product with 5 μL loading buffer (95% form amide containing 10 mmol L-1 EDTA 1:4), incubation was carried out for 10 min at 95°C and immediately for 10 min in ice, respectively and then electrophoresed on a 10% homogeneous polyacrylamide gel. The running conditions were 100 V for 16 h at 4°C. The DNA bands were visualized by silver staining.
Sequencing: PCR products showing abnormal electrophoretic mobility on SSCP gel were sequenced by Kawsar Company and analyzed by sequencing-analysis Chromas Lite 2.01 software. The sequences were compared with the wild type.
RESULTS
Present study investigate the presence of mutations, in our population of hemophilia A patients in North-Western of Iran. With the use of DNA amplification, SSCP and DNA sequencing, some mutations were detected in 35 hemophilia A patients studied. PCR amplification of a 372 bp exon 4 of the FVIII: C gene using the F8Ex4F and F8Ex4R primers, presented in Fig. 1.
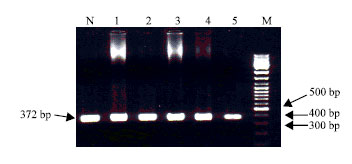 | |
| Fig. 1: | Ethidium bromide-stained 1.5% agarose gel showing PCR products corresponding to a 372 bp amplification of exon 4 using F8ex4F and F8ex4R primers. Lanes from left to right are: N: Normal male, 1, 2, 3, 4 and 5 (HA 10): Different Hemophilia A patient and M: Molecular weight standard Marker (Fermentas SM0333) |
 | |
| Fig. 2: | PCR/SSCP analysis of FVIII exon 4 in 10 hemophilia A patients. The aberrant SSCP mobility shifts in patient HA10 is marked by an arrow. (N: normal) |
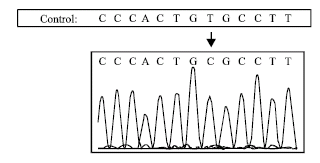 | |
| Fig. 3: | Sequencing of PCR-amplified DNA fragment from patient HA10. Comparison of the normal control sequence with the HA10 sequence indicates a base substitution of a C for a T (TGC→CGC). Point mutation is indicated by an arrow |
The mobility shift of sample HA10 was detected when the PCR products of exon 4 were analyzed by SSCP and visualized by silver-staining. This mobility shift differed from the rest of the hemophilic DNA samples and normal sample run on the same 10% homogeneous polyacrylamide gel (Fig. 2).
| Table 1: | Summary of the molecular defect in HA10 hemophilia A patient |
 | |
| *: This column gives the single letter amino-acid code for the wild-type residues in this position in (L-R): human FVIII, murine FVIII, human FV, bovine FV, rat Cp and human Cp | |
After PCR amplification and SSCP analysis, sequence analysis of PCR product showed a missense mutation at codon 153 (TGC). This missense mutation due to T → C transition at this position replaces cysteine with an arginine residue (Cysteine→Argenine) (Fig. 3). Summary of the molecular defect in HA10 hemophilia A patient is indicated in Table 1.
DISCUSSION
Identification of causative mutations in the factor VIII gene on X chromosome for families with hemophilia A can permit very accurate determination of carrier status of at-risk females in these families and provides options for prenatal diagnosis. Present research investigate the causative mutations in factor VIII gene in hemophilia A patients in North Western of Iran. To date, according to present research, some missense mutations in some exons of FVIII gene were detected, which previously have been reported. In this study, a novel missense mutation in HA10 patient was detected. This novel missense mutation due to T → C transition at codon 153 (TGC) of the factor VIII gene replaces a cysteine with an arginine residue.
The factor VIII hetero-dimer contains a heavy chain, consisting of the A1, A2 and B domains (residues 1-336, 373-740 and 741-1648, respectively) and a light chain, consisting of the A3, C1 and C2 domains (residues 1690-2019, 2020-2172 and 2173-2332, respectively). A1 subunit but not A3-C1-C2 subunit stimulates A2-dependent enhancement of Factor X conversion. The A1 subunit likely provides the primary contact for A2 subunit in factor VIIIa. The affinity for the A1-A2 interaction is approximately the same as the affinity observed for the interaction of A2 with A1/A3-C1-C2 dimer (Philip et al., 1999).
FVIII shows clear homology to FV. There is approximately 35 to 40% sequence homology. In addition the blue copper-binding plasma protein ceruloplasmin (Cp) is homologous to FVIII and FV. The three A domains of Cp show approximately 34% sequence homology with the FVIII A domains. Of the six disulphide links identified in the FVIII A domains, five (A1: 153-179, 248-329; A2: 528-554, 630-711; A3: 1832-1858) are homologous to those in hCp (Pemberton et al., 1997). Alignment of A1 domain sequences of human and murine FVIII (humf8, murf8), human and bovine FV (humfv, bovfv), rat and human Cp (ratcp, humcp), showing that the residue 153 is a conserved cysteine in all humf8, murf8, humfv, ratcp and humcp. Therefore, missense mutation due to T → C transition at this position which replaces cysteine with an arginine residue, causes deficiency in A1 domain function that result in hemophilia A. In conclusion this study has shown that the use of PCR and silver staining of SSCP techniques may be useful in detecting most of the genetic defects such as point mutations of factor VIII gene in hemophilic patients.
ACKNOWLEDGMENTS
We thank Somayeh Akrami for any helpful Laboratory cooperation and Mehdi Haggi for assistance in the early phase of this study. This research was supported by Islamic Azad University and Tabriz University.
REFERENCES
- Barry, L.S., 2002. Optimization of factor VIII replacement therapy: Can structural studies help in evading antibody inhibitors?. Br. J. Haematol., 119: 310-322.
PubMedDirect Link - Bhopale, G.M. and R.K. Nanda, 2003. Blood coagulation factor VIII: An overview. J. Biosci., 28: 783-789.
CrossRefDirect Link - Chinchang, W., V. Viprakasit, P. Pung-Amritt, V.S. Tanphaichitr and P. Yenchitsomanus, 2005. Molecular analysis of unknown β-globin gene mutations using polymerase chain reaction-single strand conformation polymorphism (PCR-SSCP) technique and its application in Thai families with β-thalassemias and β-globin variants. Clin. Biochem., 38: 987-996.
Direct Link - Sambrook, J., E.F. Fritsch and T.A. Maniatis, 1989. Molecular Cloning: A Laboratory Manual. 2nd Edn., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA., ISBN-13: 9780879695774, Pages: 397.
Direct Link