
Jacques Simpore
Centre Medical Saint Camille, 01 BP 364 Ouagadougou 01, Burkina Faso, Italy
Denise Ilboudo
University of Ouagadougou, UFR/SVT-SDS, 03 BP 7021 Ouagadougou Burkina Faso
Karou Damintoti
University of Ouagadougou, UFR/SVT-SDS, 03 BP 7021 Ouagadougou Burkina Faso
Luc Sawadogo
University of Ouagadougou, UFR/SVT-SDS, 03 BP 7021 Ouagadougou Burkina Faso
Esposito Maria
Dipartimento Clinico Sperimentale di Medicina E Farmacologia, Università di Messina, T. Biol. Policlinico Universitario, Via Consolare Valeria-Gazzi, 98125 Messina, Italy
Scott Binet
Camillian Task Force, Piazza della Maddalena, 53-00186 Roma, Italia
Henri Nitiema
Università Cattolica Sacre Cuore, Largo A. Gemelli, 8-00168 Roma, Italy
Paul Ouedraogo
Università Cattolica Sacre Cuore, Largo A. Gemelli, 8-00168 Roma, Italy
Salvatore Pignatelli
Centre Medical Saint Camille, 01 BP 364 Ouagadougou 01, Burkina Faso, Italy
Jean-Baptiste Nikiema
University of Ouagadougou, UFR/SVT-SDS, 03 BP 7021 Ouagadougou Burkina Faso
Pakistan Journal of Biological Sciences
Year: 2007 | Volume: 10 | Issue: 3 | Page No.: 409-414







ABSTRACT
Where malaria is endemic, there is an unexpected association between haemoglobinopathies and glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Their coexistence in a patient with sickle cell disease (SCD) can lead to hemolytic anemia, hemoglobinuria, sepsis, renal failure and vaso-occlusive attacks (VOA). The aim of this research was to determine the impact of G-6-PD deficiency in SCD patients. That is why, we screened haemoglobinopathies and G-6-PD deficiency in 7 villages and at 10 primary schools in Kadiogo Province, Burkina Faso. Hemoglobin electrophoresis was performed on blood from 18,383 people. From these results, we chose 342 subjects for a hemogram and the measure of the G-6-PD activity. The results were analyzed with EpiInfo-6 and Spss-10. Statistical significance was set at p<0.05. We found a prevalence of 28.9% of Sickle Cell Trait (SCT), 1.3% of Major Sickle Cell Syndromes (MSCS), 12.3% of G-6-PD deficiency among women and 20.5% among men. We did not detect a statistically significant difference for counts of erythrocytes (p = 0.773), leucocytes (p = 0.227) and reticulocytes (0.292); hemoglobin levels (p = 0.998); annual vaso-occlusive attacks (p = 0.869) between persons with SCD having a G-6-PD deficiency and those with normal G-6-PD activity. According to this study, G-6-PD deficiency does not seem to increase the severity of SCD. However, these patients should know their G-6-PD genotype in order to avoid consuming oxidative drugs that might provoke oxidative stress.
PDF Abstract XML References Citation
How to cite this article
Jacques Simpore, Denise Ilboudo, Karou Damintoti, Luc Sawadogo, Esposito Maria, Scott Binet, Henri Nitiema, Paul Ouedraogo, Salvatore Pignatelli and Jean-Baptiste Nikiema, 2007. Glucose-6-Phosphate Dehydrogenase Deficiency and Sickle Cell Disease in Burkina Faso. Pakistan Journal of Biological Sciences, 10: 409-414.
DOI: 10.3923/pjbs.2007.409.414
URL: https://scialert.net/abstract/?doi=pjbs.2007.409.414
DOI: 10.3923/pjbs.2007.409.414
URL: https://scialert.net/abstract/?doi=pjbs.2007.409.414
INTRODUCTION
Burkina Faso, a country in Western Africa, is bordered on the north and the west by Mali, on the east by Niger and the south by Ivory Coast, Ghana, Togo and Benin. In tropical area, where malaria is endemic, there is a high prevalence of haemoglobinopathies and G-6-PD deficiency (Enevold et al., 2005; Fleming et al., 1979). The genes for sickle cell disease (SCD) and glucose-6-phosphate dehydrogenase (G-6-PD) deficiency are located on chromosome 11 and chromosome X, respectively. During cell division, the genes normally assort independently (Bouanga et al., 1998; Luzzatto et al., 1968). Sickle cell patients experience vaso-occlusive attacks (VOA), bacterial infections, priapism and chronic visceral complications associated with ischemia in different organs (Jacob et al., 2005. Singh et al., 2006. Rogers, 2005). Major Sickle Cell Syndromes (MSCS) are the most prevalent forms of genetic disease in the wider malaria area in tropical countries. Glucose-6-phosphate dehydrogenase deficiency also occurs with increased frequency throughout Africa, Asia, the Mediterranean and the Middle East. In G-6-PD deficient individuals, anemia is usually caused by fava beans (Laosombat et al., 2006) and other oxidative drugs (Ciftci, 2005; Ozmen et al., 2004): aspirin, primaquine and quinine that are used during malaria attacks. In addition to hemolytic anemia, G-6-PD deficient individuals are predisposed to prolonged neonatal jaundice, a result of neonatal hyperbilirubinemia (Muzaffer, 2005; Kaplan et al., 2005). This is a potentially serious problem as neonatal hyperbilirubinemia can cause severe neurological complications and even death (Diop et al., 2005). The high prevalence of hemoglobin S (HbS) and G-6-PD deficiency (A-type) in tropical areas is related to the selective advantage they provide against malaria (Williams et al., 2005; Shimizu et al., 2005). The reported prevalence of G-6-PD deficiency in SCD patients varies. According to some authors, the incidence of G-6-PD deficiency is higher in MSCS patients than the general population (Bouanga et al., 1998; Lewis et al., 1966; Samuel et al., 1986). Other authors do not agree with this association believing rather that the mutated genes responsible for these states do not stay on the same chromosome, so they cannot be linked (Bouanga et al., 1998; Luzzatto et al., 1968). The potential influence of glucose-6-phosphates dehydrogenase deficiency upon the clinical manifestation of patients with SCD is a matter of contention as well (El-Hazmi et al., 1987; Bienzle et al., 1975). It is known, however, that G-6-PD deficiency in patients with SCT can express itself like symptomatic SCD. Screening of SCD patients who are carrying the G-6-PD deficiency gene will guide physicians in their care. Such measures allow sickle cell disease patients to enjoy a life expectancy beyond 50 years. This study has three aims: i) to estimate the prevalence of the association between the haemoglobinopathies and G-6-PD deficiency genes among the population, ii) to look at patients who carry this double mutation and to determine the evolution of any hematological parameters that might be therapeutically helpful, iii) finally, to see if this curious linkage of these two mutated genes causes more complications in the pathogenesis of SCD patients.
MATERIALS AND METHODS
Blood samples were collected in 7 villages and at 10 primary schools of the Province of Kadiogo in Burkina Faso from May 25, 1999 to June 15, 2005. A total of 18,383 subjects, ages 7-32 years, average 21.81±8.47 years, were enrolled in the SCD study. From the electrophoresis of hemoglobin results, we chose 342 individuals for the following analyses: hemogram and G-6-PD test. All the SCD patients agreed to answer questions concerning their health status: number of annual transfusions, hospitalizations, vaso-occlusive attacks (VOA) and episodes of malaria. We compared the different evaluative elements of SCD patients who have G-6-PD deficiency with SCD patients who do not have a G-6-PD deficit. We calculated a severity index, a total equal to the sum of the scores obtained utilizing the following parameters:
Number of hospitalizations in the year: 0 times at score = 0; 1 to 2 times at score = 1; more than 2 times at score = 2
Number of vaso-occlusive attacks (VOA) in the year: 0 times at score = 0; 1 to 2 times at score = 1; more than 2 times at score = 2
Number of transfusions in the year: 0 times at score = 0; 1 to 2 times at score = 1; more than 2 times at score = 2
Number of malaria attacks in the year: 0 times at score = 0; 1 to 2 times at score = 1; more than 2 times at score = 2
Number of suspensions of activity in the year: 0 times at score = 0; 1 to 2 times at score = 1; more than 2 times at score = 2
Ethical committee: The Ethics Committee of Saint Camille Medical Center in Ouagadougou approved the admission of prospective participants into the study only after informed consent was obtained.
Methods: Ten milliliter of venous blood was drawn from each person and placed in two EDTA tubes. One blood tube was used soon thereafter for the hemoglobin electrophoresis and hematological parameters. Within 3 h of drawing blood, plasma was separated out by centrifugation at 3,000 rpm for 10 minutes in the 2nd tube to be used for the G-6-PD test. All subjects underwent hemoglobin electrophoresis using cellulose acetate plates (Helena Laboratories, Beaumont, TX, the USA) with a pH 8.6 buffer. A hemogram was carried out utilizing the Cell-dyn®1700 automat of Abbott House while reticulocytes were measured using new methylene blue dye. Glucose-6-phosphate dehydrogenase quantities were measured by Quantitative G-6-PD Diagnostic Real Time Systems Kit Test (Italy) using a spectrophotometer reader type Microlab 2000.
Statistical analysis: Demographic and clinical profiles were recorded on computer files and analyzed by standard software SPSS-10 and EpiInfo-6. Statistical significance was set at p<0.05.
RESULTS
Hemoglobin electrophoresis was performed on a sample of 18,383 individuals: 9,923 females (53,98%) and 8,460 males (4600%). Table 1 presents allelic frequencies calculated utilizing the genotypes observed: p, q and r are respectively the frequency of βA, βc and βs. The formula of Hardy-Weinberg allowed us to calculate the genotypic and allelic frequencies: N(p+q+r)2: AA = Np2; AC = 2Npq; AS = 2Npr; CC = Nq2; SC = 2Nqr; SS = Nr2. N indicates the number of the sample. Table 2 presents the G-6-PD genotypic frequencies according to age groups. The frequency of G-6-PD deficiency in the general population is 16.3%. This percentage increases with age: 6-9 years (8.4%), 10-19 years (20.9%) and 20-29 years (25.0%). Table 3 shows G-6-PD activity according to hemoglobin genotype after hemoglobin electrophoresis. The frequency of G-6-PD deficiency among SCT, MSCS and the control population with Hb AA are, respectively: 18.57%, 27.0% and 7.81%. It is important to note that the prevalence of the favism is higher in men (20.5%) than women (12.3%), with p = 0.041. Table 4 presents the results of the hemogram and the G-6-PD activity of AA, SCT and MSCS subjects. The t-TEST reveals a statistically significant difference (p<0.001) between the control population with Hb AA and MSCS patients for G-6-PD enzymatic activity, the number of red blood cells (RBCs), white blood cell (WBCs), reticulocytes, the concentration of hemoglobin and the mean corpuscular volume (MCV). Whereas the control population with Hb AA presents a normal hemogram, there is a severe anemia, increased amount of reticulocytes and leucocytes associated with MSCS subjects. The SCT subjects have a moderate anemia and reticulocytosis. Table 5 presents the influence of the G-6-PD deficiency on the pathogenesis of SCD. In our samples, we had 74 SCD patients: 37 hemoglobin homozygote SS and 37 hemoglobin double heterozygote SC. Among these patients, 54 had normal G-6-PD activity and 20 had deficient G-6-PD activity. The subjects freely answered the questionnaire, the results of which are reflected in Table 5.
| Table 1: | Genotype and allelic frequencies observed and calculated |
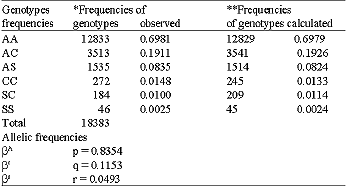 | |
| NS = Not significant. χ2 *---> **: p = 0.659 (NS) | |
| Table 2: | G-6-PD genotypic frequencies according to age group |
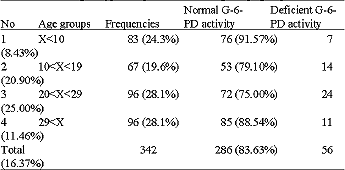 | |
χ2 Test, 1-2: p = 0.029, 2-3: p = 0.542 (NS), 1-3: p = 0.003, 2- 4: p = 0.099 (NS), 1-4: p = 0.502 Not Significant (NS), 3-4: p = 0.015 | |
| Table 3: | G-6-PD frequencies and hemoglobin genotype distribution |
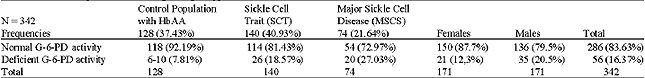 | |
χ2 Test: AA/G-6-PD-_ SCT/G-6-PD-: p = 0.010; AA/G-6-PD-_ MSCS/G-6-PD-: p<0.001; SCT/G-6-PD-_ MSCS/G-6-PD-: p<0.152 (NS), Not Significant | |
| Table 4: | G-6-PD activity and Hemogram parameter distribution |
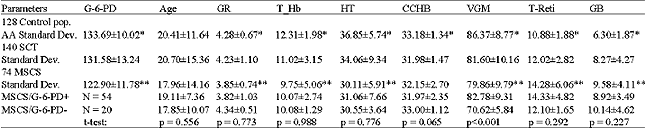 | |
| * t-test: p<0.001; ** t-test: p<0.04 | |
| Table 5: | G-6-PD deficit and SCD pathogenesis score |
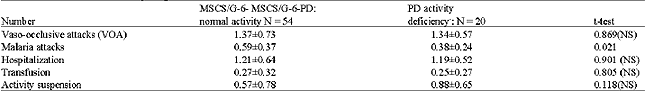 | |
DISCUSSION
For the 18,383 samples from the 10 schools of the town of Ouagadougou and the 7 surrounding villages of the capital, we found 5,320 (28,9%) SCT (HbAS, HbAC, HbCC) and 230 (1.3%) MSCS (HbSS and HbSC) (Table 1). We discovered 19.1% of the haemoglobinopathy AC. This frequency is higher than that identified in Mali (15.8%) (Diallo et al., 1994) and in Nigeria (0.7%) (Storey et al., 1979). According to Trabuchet (1991) the epicenter of the HbC mutation could be in West Africa. Indeed, studying the haemoglobinopathies among the Mossi in Burkina Faso, Modiano et al. 2001a discovered that hemoglobin CC protected against severe malaria. In this research, we found AS in 8.4% of subjects. This prevalence is lower than that detected in Ghana 14,0% (Mockenhaupt et al., 2000), Dhelki Kharia (India) 12.5% (Balgir, 2005), but very similar to that detected in Paik (India) 7.4% (Balgir, 2005), in Karachi (Pakistan) 5.1%, (Ghani et al., 2002). Our result was quite higher than that found in Paraja (India) 0.9% (Balgir, 2005) and in Upper Volta 4.9% by Labie, (1984). The slight increase in the HbS percentage in our sample compared with that of Labie in (1984) is certainly due to the additional medical care provided in Burkina Faso since 1990 for SCD patients: vaccinations against pneumococcus, antimicrobial prophylaxis and improved medical care. Simpore et al. (2002) with their haemoglobinopathy research, demonstrated that SCD patients are often sick and frequently go for medical consultation at Saint Camille Medical Centre in Ouagadougou: 660 HbSC (6.5%), 196 HbSS (1.9%) and 3400 SCT (33.4%). We detected 16.37% G-6-PD deficiency (Table 2). This prevalence is higher than that they found in Dakar in 2005 (12.3%) (Diop et al., 2005), in Pakistan (1.8%) (Ali et al., 2005) and lower than that detected in both Lome in 2001 (24,1%) (Gbadoe et al., 2001) and Vataliya Prajapati community (India) (22,0%) (Gupte et al., 2005). However, our G-6-PD deficiency frequency is almost the same as Modiano et al. (2001a) identified (14.9%) (Modiano et al., 2001b). The prevalence of G-6-PD deficiency in male subjects (20.5%) differs significantly (p = 0.04) from that found in women (12.3%) Table 3. That is due to the fact that the males are hemizygous and the females are dizygous for X chromosome. As such, the probability of finding the two genes for the G-6-PD mutations on the chromosome X is lower. In this study we found a very slight but significant difference of G-6-PD enzyme activity between male (121.2±13.2 mU.109/Eryth) and female (123.9±12.1 mU.109/Eryth) p<0.05. This similarity of G-6-PD enzymatic activity between male and female is due to the fact that the phenomenon of lionization activates only one X chromosome in each woman`s cell. G-6-PD deficiency and SCD are two genetic disorders of red blood cells (RBCs) which predispose to hemolytic anemia. Their presence in patients should lead those providing medical care to avoid using oxidative drugs. In our research we found a significant difference between G-6-PD deficiency in SCD patients (27.03%) and in the control population with HbAA (7.81%) p<0.001 (Table 3). Similar results were found in Congo Brazzaville (Samuel et al., 1986), in Senegal (Diop et al., 2005), in Kenya (Rattazzi et al., 1971), in Ghana (Lewis, et al., 1973) and in Turkey (Akoglu et al., 1986). But other studies do not confirm this association (El-Hazmi et al., 1987; Steinberg and Dreiling, 1974; Saad et al., 1992). More often than not, at the time of meiosis during the cycle of cell division, the HbS and G-6-PD deficiency genes, which are located on two different chromosomes, independently segregate according to Mendel`s Law. But the phenomenon of selection, due to the presence of plasmodia, pushes these pathogenic genes to stay together and thus associated. Comparing the profile of SCD subjects with G-6-PD deficiency and SCD subjects with normal G-6-PD activity, no statistical difference was found between these two groups with regard to annual VOAs (p = 0.869), annual hospitalizations (p = 0.901), annual transfusion frequency (p = 0.805) and annual activity suspension (0.118) (Table 5). Similarly, no statistical difference was found for hematological parameters: RBC counts (p = 0.773), reticulocyte counts (0.292), WBC counts (p = 0.227), hemoglobin level (p = 0.988), hematocrit (p = 0.776) and mean corpuscular Hb (MCH) (p = 0.065). We identified a statistical difference between the two groups only for MCV values (p<0.001). These results agree with the studies of Bienzle et al. (1975), Steinberg,and Dreiling. (1974), Diop. (2000) and El-Hazmi. (1986), which showed that G6PD deficiency, even in the tropical area, seems to have no effects on the hematological data and clinical evolution of HbSS patients. According to these authors, G6PD deficiency does not offer any advantage or disadvantage to patients with sickle cell disease. However, other authors like Bouanga. (1998) and Znaidi. (1995) offer different results. And some authors argue that G-6-PD deficiency has a protective effect in SCD patients because the absence of enzymatic activity contributes to the elimination of premature RBCs. There is a elevated generation of young erythrocytes in these G-6-PD deficiency persons (Steinberg and Dreiling, 1974). There was a significant difference in malaria attack frequency between the group of SCD patients with G-6-PD deficiency and SCD subjects with normal G-6-PD activity (p = 0.048). SCD subjects who have a G-6-PD deficiency suffer fewer crises of malaria. It is known that the frequency of the G-6-PD deficit increases with age (Table 2), which could indicate a longevity advantage in malaria zones as compared to people with normal enzyme activity. Nonetheless, these results should not lead us to forget that when SCD patients with G-6-PD deficiency have malaria, there is a significant risk of developing hemolytic anemia crises, sepsis, hemoglobinuria and renal failure after taking anti-malarials or eating oxidative foods like fava beans. SCD, which is responsible for a high rate of medical consultations and increased mortality in children, remains a public health problem for tropical countries. It remains vital to screen for G-6-PD deficiency among SCD patients in order to provide appropriate preventive and therapeutic measures.
ACKNOWLEDGMENTS
The authors want to thank all the villages and schools that agreed to take part in this study and the laboratory technicians of St. Camille Medical Centre, Ouagadougou. In particular, we are grateful for the skillful help of Mr. Bakamba Robert, Mme. Justine Yara, Mr. Emmanuel Bouda and Mme. Angèle Sanfo Bambara. This research would not have been possible without the friendly and constructive support of the Italian Episcopal Conference (C.E.I.) and the RADIM House in Italy (Doctor Luigi SPARANO).
REFERENCES
- Ali, N., M. Anwar, M. Ayyub, F.A. Bhatti, M. Nadeem and A. Nadeem, 2005. Frequency of glucose-6-phosphate dehydrogenase deficiency in some ethnic groups of Pakistan. J. Coll. Phys. Surg. Pak., 15: 137-341.
Direct Link - Balgir, R.S., 2005. The spectrum of haemoglobin variants in two scheduled tribes of Sundargarh district in northwestern Orissa, India. Ann. Hum. Biol., 32: 560-573.
Direct Link - Ciftci, M., 2005. Effects of some drugs on the activity of glucose 6-phosphate dehydro-genase from rainbow trout erythrocytes in vitro. J. Enzyme Inhib. Med. Chem., 20: 485-489.
Direct Link - Diop, S., A. Sene, M. Cisse, A.O. Toure and O. Sow et al., 2005. Prevalence and morbidity of G6PD deficiency in sickle cell disease in the homozygote. Dakar Med., 50: 56-60.
Direct Link - Enevold, A., L.S. Vestergaard, J. Lusingu, C.J. Drakeley and M.M. Lemnge et al., 2005. Rapid screening for glu-cose-6-phosphate dehydrogenase deficiency and haemoglobin polymorphisms in Africa by a simple high-throughput SSOP-ELISA method. Malar. J., 4: 4-61.
CrossRefDirect Link - Gbadoe, A.D., K.Atsou, O.A. Agbodjan-Djossou, E. Tsolenyanu and M. Nyadanu et al., 2001. Ambulatory management of sickle cell disease: Evaluation of the first year follow up of patients in the pediatric department of Lome, Togo. Bull. Soc. Pathol. Exot., 94: 101-105.
Direct Link - Ghani, R., M.A. Manji and N. Ahmed, 2002. Hemoglobinopathies among five major ethnic groups in Karachi, Pakistan. Southeast Asian. J. Trop. Med. Public Health, 33: 855-861.
Direct Link - Gupte, S.C., P.U. Patel and J.M. Ranat, 2005. G6PD deficiency in vataliya prajapati community settled in Surat. Ind. J. Med. Sci., 59: 51-56.
Direct Link - Jacob, E., J.E. Beyer, C. Miaskowski, M. Savedra, M. Treadwell and L. Styles, 2005. Are there phases to the vasoocclusive painful episode in sickle cell disease? J. Pain Symptom Manage., 29: 392-400.
Direct Link - Kaplan, M., M. Muraca, H.J. Vreman, C. Hammerman and M.T. Vilei et al., 2005. Neonatal bilirubin production-conjugation imbalance: Effect of glucose-6-phosphate dehydrogenase deficiency and borderline prematurity. Arch. Dis. Child Fetal Neonatal Ed., 90: F123-F127.
Direct Link - Laosombat, V., B. Sattayasevana, T. Chotsampancharoen and M. Wongchanchailert, 2006. Glu-cose-6-phosphate dehydrogenase variants associated with favism in Thai children. Int. J. Hematol., 83: 139-143.
Direct Link - Mockenhaupt, F.P., B. Rong, M. Gunther, S. Beck and H. Till et al., 2000. Anaemia in pregnant Ghanaian women: Importance of malaria, iron deficiency and haemoglobinopathies. Trans. R. Soc. Trop. Med. Hyg., 94: 477-483.
CrossRefDirect Link - Modiano, D., G. Luoni, B.S. Sirima, J. Simpore and F. Verra et al., 2001. Haemoglobin C protects against clinical Plasmodium falciparum malaria. Nature, 414: 305-308.
CrossRefDirect Link - Modiano, D., G. Luoni, B.S. Sirima, A. Lanfrancotti and V. Petrarca et al., 2001. The lower susceptibility to Plasmodium falciparum malaria of Fulani of Burkina Faso (West Africa) is associated with low frequencies of classic malaria-resistance genes. Trans. R Soc. Trop. Med. Hyg., 95: 149-152.
Direct Link - Muzaffer, M.A., 2005. Neonatal screening of glucose-6-phosphate dehydrogenase deficiency in Yanbu, Saudi Arabia. J. Med. Screen., 12: 170-171.
CrossRefDirect Link - Ozmen, I. and O.I. Kufrevioglu, 2004. Effects of antiemetic drugs on glucose 6-phosphate dehy-drogenase and some antioxidant enzymes. Pharmacol. Res., 50: 499-504.
Direct Link - Shimizu, H., M. Tamam, A. Soemantri and T. Ishida, 2005. Glucose-6-phosphate dehydrogenase deficiency and Southeast Asian ovalocytosis in asymptomatic Plasmodium carriers in Sumba island, Indonesia. J. Hum. Genet., 50: 420-424.
Direct Link - Simpore, J., S. Pignatelli, S. Barlati and S. Musumeci, 2002. Biological and clinical presentation of patients with hemoglobinopathies attending an urban hospital in Ouagadougou: Confirmation of the modification of the balance between HbS and HbC in Burkina Faso. Hemoglobin, 26: 121-127.
Direct Link - Singh, S., D.K. Singh, R. Gupta, S. Nigam and T. Singh, 2006. Persistent splenomegaly in an adult female with homozygous sickle cell anemia. Hematology, 11: 63-65.
Direct Link - Trabuchet, G., J. Elion, O. Dunda, C. Lapoumeroulie and R. Ducrocq et al., 1991. Nucleotide sequence evidence of the unicentric origin of the beta C mutation in Africa. Hum. Genet., 87: 597-601.
Direct Link - Williams, T.N., T.W. Mwangi, S. Wambua, N.D. Alexander and M. Kortok et al., 2005. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. J. Infect. Dis., 1: 178-186.
Direct Link