
Christine Cheryl Fernandez
Laboratory of Enzyme and Microbial Technology, Institute of Bioscience, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
Noor Aini Abdul Rashid
Department of Biology, Faculty of Science, Universiti Teknologi, Malaysia
Zaharah Ibrahim
Department of Biology, Faculty of Science, Universiti Teknologi, Malaysia
Piakong M. Tuah
Department of Biology, Faculty of Science, Universiti Teknologi, Malaysia
Pakistan Journal of Biological Sciences
Year: 2006 | Volume: 9 | Issue: 5 | Page No.: 805-809







ABSTRACT
In this study, RETL-Cr1 was studied for its growth and degradation properties in M3 and modified Ramsay media and in the latter with varying saline concentrations. There was no significant effect of medium salinity towards the growth and degradation of phenol in RETL-Cr1. From the growth profile, μ was calculated to be 0.288 h-1 while td was 2.971 h. The significantly higher intracellular enzyme activity of the crude extract was assayed against varying pHs, ethylenediaminetetraacetic acid (EDTA) concentrations, temperatures and Nicotinamide adenine Dinucleotide (NADH) concentrations, in that order with the resulting optimized conditions of pH 6.5, 1.8 mM EDTA, 37°C and 0.4 mM NADH. Enzyme stability was assayed against varying pH and temperature where it was most stable at pH 6.5 and between temperatures ranging from 25 and 30°C. The crude phenol degrading enzyme was further subjected to kinetic studies at the optimized conditions to determine its affinity towards phenol at varying concentrations. From the Lineweaver-Burk plot it was found that the crude enzyme has a high Vmax value of 4.963 μM phenol degraded per minute and a low Km value of 2.115 μM suggesting high affinity towards the substrate. Confirmation of phenol degradation intermediate was determined upon the presence of catechol via thin layer chromatography.
PDF Abstract XML References Citation
How to cite this article
Christine Cheryl Fernandez, Noor Aini Abdul Rashid, Zaharah Ibrahim and Piakong M. Tuah, 2006. Development of an Enzyme Assay and Preliminary Kinetic Studies for the Enzyme (s) from Candida troplicalis RETL-Crl Involved in Phenol Degradation. Pakistan Journal of Biological Sciences, 9: 805-809.
DOI: 10.3923/pjbs.2006.805.809
URL: https://scialert.net/abstract/?doi=pjbs.2006.805.809
DOI: 10.3923/pjbs.2006.805.809
URL: https://scialert.net/abstract/?doi=pjbs.2006.805.809
INTRODUCTION
Until recently, studies on biodegradation of phenolic compounds which are the major constituents of wastewaters from petrochemical, chemical and steel industries have received much attention as the primary form of environmental remediation complementing the physical and chemical methods (Komarkova et al., 2003; Chang et al., 1998). Ecologists have come to realize the increasing risk o f phenolic compounds and its by-products as pollutant to the environment especially water reservoirs which may lead to imminent integration of phenol in the food chain (IPCS, 1994).
As a typical xenobiotic, phenol solutions are corrosive to the skin and eyes and particularly lethal to humans if consumed at a minimal amount of 4.8 g. Also known as hydroxybenzene, phenol is a component of coaltar and petrochemical compounds. Formed during the natural decomposition of organic materials it contributes to the rising environmental levels via forest fires (Hubble et al., 1981) and irresponsible wastewater disposal. However it is susceptible to photolysis. Being rather sensitive to oxidizing agents, in the environment, phenol reacts with photochemically produced hydroxyl and peroxy radicals in sunlit natural waters. It is oxidized when the hydrogen atom is split from the hydroxyl group followed by resonance stabilization of the phenyloxy radical. The phenyloxy radical will be further oxidized depending on the oxidizing agent and reaction condition (IPCS, 1994). Mineralization occurs when it is oxidized to carbon dioxide and water at 50°C in the presence of oxygen and sunlight at a rate of 11% in 24 h (Howard, 1989).
Eukaryotes and prokaryotes also evolved their own mechanism for phenol and phenolic compounds degradation by incorporating it in the main metabolic cycle. It has been extensively studied on the basis of phenol degradation at low concentrations but not at high concentrations (Ruiz-Ordaz et al., 2001). Being the most widely studied eukaryote that degrades phenol and its derivatives at a concentration of up to 3000 ppm, Candida tropicalis is classified as a chemolithotroph since it is able to thrive solely on phenol as its main carbon and energy source. The phenol degradation in C. tropicalis is catalyzed mainly by phenol hydroxylase and catechol-1, 2-dioxygenase via the ortho ring cleavage pathway which is also known as the β-ketoadipate pathway of catechol to cis, cis-muconic acid (Bastos et al., 2000a,b). Both are inducible enzymes and function in the presence of oxygen (Chang et al., 1998). The end products of phenol degradation in eukaryotic cells were succinic acid and acetyl CoA (Feist and Hegeman, 1969). This acetyl CoA initiated the TCA or Krebs cycle which is one of the main metabolic pathways and succinic acid is an important constitute of this cycle (Harwood and Parales, 1996).
Bastos et al. (2000a) found that the highly versatile C. tropicalis was able to grow and degrade higher concentrations of phenol at higher medium salinity at an even faster rate as compared to its native environment. Thus it was of our interest to determine the effect of medium salinity towards the growth and phenol degradation rate. The efficiency of the cell fractions in degrading phenol was also assayed particularly the crude cell extract which was assayed and optimized against several parameters.
MATERIALS AND METHODS
This study was carried out between November 2004 and February 2005.
Candida tropicalis RETL-Cr1 was isolated from local petrochemical wastes of the shores of Johor and Negeri Sembilan, Malaysia. It was found to be the best phenol degrader of four other isolates, ideally cultured in Ramsay medium. Therefore, it is of our interest to determine the presence of phenol degrading enzymes by developing a suitable enzyme assay. The enzymes made up a concoction of preliminary enzymes responsible for the metabolism of phenol in RETL-Cr1.
Phenol standard curve and the determination of the phenol concentration in samples were based upon the colourimetric method introduced by Box (1983) with slight modifications with the absorbance being read at 765 nm. The determination of protein concentration was carried out according to the method established by Lowry et al. (1951) with modifications to accommodate this study.
Ramsay (MRM) and M3 media were prepared to monitor the growth and phenol degradation rates in both the media with appropriate modifications to accommodate this study. MRM contained the chemical reagents described by Ramsay et al. (1983). 2 g L-1 NH4NO3 (Fluka), 0.5 g L-1 KH2PO4 (Merck), 1 g L-1 K2HPO4 (Sigma), 0.1 g L-1 KCl (Riedel de Haen), 0.06 g L-1 yeast extract (Oxoid), 2 g L-1 NaCl (Merck) and stock solutions of MgSO4 (GCE) and CaCl2 (GCE) both at 10 g L-1 were prepared separately and autoclaved before being added to a final concentration of 0.5 and 0.01 g L-1, respectively.
M3 medium was prepared by preparing stock solutions of MgSO4 (GCE), 10 g L-1 and of NaCl (Merck), 100 g L-1 which was autoclaved separately before adding to the other sterilized components which were NH4NO3 (Fluka) 1.0 g L-1, KH2PO4 (Merck) 0.5 g L-1, K2HPO4 (Sigma) 0.5 g L-1, KCl (Riedel de Haen) 0.1 g L-1 and trace minerals H3BO3 0.5 g L-1, CuSO4 1.0 g L-1, KI 1.0 g L-1, MnSO4 2.0 g L-1, ZnSO4 5.0 g L-1. The final phenol concentration in both the MRM and M3 media was 3 mM. Only the MRM was used in determining the effect of medium salinity (NaCl) at the range of 0-8 g L-1. Long term glycerol stock culture was prepared and stored at –80°C.
The assay components which were phenol (0.2 mM), NADH (0.1 mM), Na2EDTA (1.0 mM) and phosphate buffer (0.1 mM) solutions were prepared as a standard initial reaction mixture. This standard initial reaction mixture was used in determining enzyme localization and the enzyme activity of the various cell fractions (Rosenberg, 1996; Scopes, 1994). The assay was initiated upon addition of the cell fractions (500 μL) and NADH. The enzyme activity (U) can be defined as 1 μM of compound being formed or consumed per minute. In this study the U was defined as μM of phenol being degraded per minute. In order to obtain the crude cell free extract, the yeast cells obtained during the maximum log phase were subjected to the glass bead grinding method (Rosenberg, 1996). The other cell fractions include the resting cells, cell debris and the culture supernatant.
Optimization of the pH, EDTA concentration, temperature and NADH concentration were carried out in that respective order with suitable control experiments. Determination of enzyme stability against varying pH and temperature were done subjecting the assay to extended incubation time. The optimized conditions were employed in the final assay to determine the Km and Vmax of the crude enzyme. TLC was done to confirm the intermediary products of phenol degradation (Leow, 2004).
RESULTS AND DISCUSSION
Results obtained showed that RETL-Cr1 grew and degraded phenol more significantly in MRM rather than in M3 medium. The lag phase of growth and phenol degradation rate of RETL-Cr1 in MRM was significantly shorter than in M3. This shows that the M3 medium was not preferable for the growth and degradation of phenol by RETL-Cr1. Figure 1 shows the growth profile and growth associated phenol degradation. The RETL-Cr1 specific growth rate (μ) was 0.233 h-1 with the doubling time (td) of 2.971 h in MRM.
Interestingly, no inhibitory effect on growth was observed with no acceleration in either growth or phenol degradation. The results indicate that low saline concentration did not have any significant effect on growth and phenol degradation (data not shown).
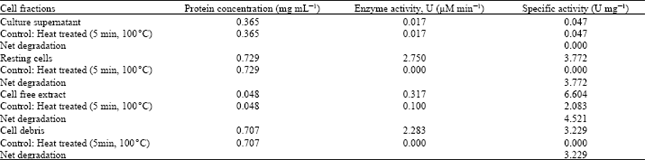
| Table 1: | Specific activity for cell fractions |
 | |
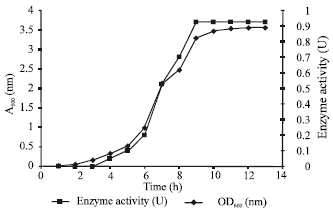 | |
| Fig. 1: | Growth associated phenol degradation |
Present results are contrasting to Bastos et al. (2000a) who reported that repeated adaptation phases of C. tropicalis on saline media was important to allow adaptation of its phenol regulatory system in order that the adapted microorganism have higher phenol tolerance and degrade higher phenol concentrations.
Table 1 shows that cell free extract which was the crude intracellular enzyme had the highest specific activity relative to resting cells, cell debris and culture supernatant. A high possibility could be that the phenol degrading enzymes are membrane bound due to the significant enzyme activity from the cell debris and resting cells fractions. This possibility was also expressed by Bastos et al. (2000b) by stating that there could be membrane level phenol degradation in Candida tropicalis.
It was found that the most suitable incubation time for the enzyme assay was 30 min using the stop enzyme assay method (Scopes, 1994). The incubation period of 30 min in comparison to the 60 min yielded a higher enzyme activity since the amount of phenol degraded was divided by the duration of the assay. The data obtained was a mean of triplicates. The reaction progress followed a parabolic curve wherein it deviated from the true initial steady-state linear rate which was distinctive when using the stop method. Proper measures should be observed in order to avoid underestimating the initial steady-state and to impose the assumption of linearity.
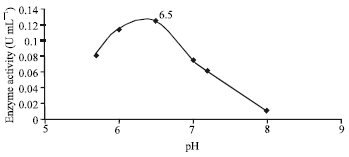 | |
| Fig. 2: | Effect of pH on enzyme activity |
Linearity can be monitored by carrying out a series of analyses at different incubation times and to detect any sequential side reactions which may be done by removing the measured product (Scopes, 1994). Usually, the initial rate phase of an enzymic reaction persists for 10 sec up to several hundred seconds which is especially rapid (Purich, 1983).
Optimal enzyme activity was observed at pH 6.5 (Fig. 2) wherein it was most stable. Significant steady decrease in enzyme activity was observed as the pH increased to the alkaline range up to pH 8. However, the enzyme showed salient activity within a wide range of pH. The results are in good agreement with that of Hannaford and Kuek (1999) and Mordocco et al. (1999) who observed that slightly acidic conditions resulted in heightened rates of phenol degradation. Extreme pHs showed lower activity due to protein denaturation and inactivation of the active sites (Scopes, 1994). The presence of EDTA greatly increased the enzyme activity with an optimal of 1.8 mM. The assay without EDTA yielded no enzyme activity. This showed that the enzyme is heavily dependent on the presence of EDTA in order to quench the inhibitory effect of the metalloproteinases found abundantly in crude yeast extracts (Suelter, 1985).
The optimum temperature for the highest enzyme activity was 37°C (Fig. 3), however, it was most stable at 25°C. Significant enzyme activity concentrated between a very narrow range of temperatures between 35 and 40°C.
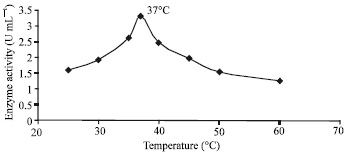 | |
| Fig. 3: | Effect of temperature on enzyme activity |
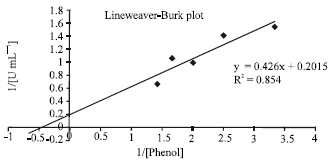 | |
| Fig. 4: | Double reciprocal Lineweaver-Burk plot to determine optimal substrate concentration under optimal conditions |
It was noted that the enzyme activity deteriorated considerably as the temperature rose from 45°C onwards. The stability of the enzyme decreased dramatically as the assay temperature rose from ambient to physiological and became extremely unstable as the temperature increased from 37 to 50°C. This was also due to conformational changes in the protein structure or denaturation of the catalytic site that led to enzyme inactivation (Scopes, 1994). Regardless of the enzyme properties, inactivation effects of both temperature and pH on the enzyme conformation should be given much consideration since both are closely related. This is because the ionization constants of protein groups may vary with temperature and in turn will affect the pH values of the buffer solutions (Scopes, 1994; Suelter, 1985).
NADH is an important cofactor especially for intracellular enzymes such as the phenol degrading enzyme from RETL-Cr1. It was observed that enzyme activity was optimal at 0.4 mM of NADH with a wide range of salient enzyme activity spanning from 0.13 to 0.53 mM. As with most intracellular enzymes which are holoenzymes, the phenol degrading enzyme too needed a cofactor as an activator such as NADH or NADPH as the primary electron donor (Madigan et al., 2000). The cofactor NADH contributes an electron to the phenol degrading system in the form of a hydride ion. Being an intracellular enzyme and a key enzyme in the biodegradation of phenol, phenol hydroxylase activity in Candida tropicalis essentially depends on the presence of NADPH (Neujahr et al., 1974). The oxidation and reduction of NADH during intracellular metabolism in yeast is potentially due to the presence of an NADH oxidoreductase system (Scopes, 1994; Suckling and Gibson, 1998).
The Michaelis-Menten constant (Km) calculated from the Lineweaver-Burk plot in Fig. 4 showed a low value of 2.115 μM depicting a relatively high affinity of the enzyme towards its substrate phenol. The maximum velocity or maximum activity (Vmax) of the enzyme was calculated to be 4.963 μM of phenol degraded per minute. There may exist a slight substrate inhibition at high phenol concentrations, thus, the substrate concentration should be reduced to about 2 to 3 times Km value or lower.
Comparison done using TLC with catechol and cis,cis muconic acid standards confirmed that intermediary products of phenol degradation which are catechol and cis,cis muconic acid were indeed present in the reaction mix following termination of the assay (Leow, 2004; Wade, 1995).
From this study it can be concluded that growth and phenol degradation rate of Candida tropicalis RETL-Cr1 were not affected by slight medium salinity. The phenol degrading enzymes were mostly intracellular and also membrane bound which for the former the enzyme assay has been successfully optimized with the Km and Vmax of 2.115 and 4.963 μM, respectively. Extensive work has yet to be done in order to obtain pure and viable phenol degrading enzymes which are primarily phenol hydroxylase and catechol-1,2-dioxygenase. Partial purification and further characterization of the enzymes should be undertaken. Molecular methods via PCR and DNA sequencing may also be implemented to obtain the DNA sequence of the designated protein responsible for phenol degradation. These genes may then be inserted into compatible plasmid vector which in turn may be transformed into E. coli. Coupled with the manipulation of gene expression, mass excretion of the designated enzymes can be obtained for further research. Alternative methods for monitoring enzyme activity should be implemented and continuous enzyme assay may be tested.
REFERENCES
- Hannaford, A.M. and C. Kuek, 1999. Aerobic batch degradation of phenol usingiImmobilized Pseudomonas putida. J. Indust. Microbiol. Biotechnol., 22: 121-126.
Direct Link - Komarkova, E., J. Paca, E. Klapkova, M. Stiborova, C.R. Soccol and M. Sobotka, 2003. Physiological changes of Candida tropicalis population degradating phenol in fed batch reactor. Brazilian Arch. Biol. Technol., 46: 537-543.
Direct Link - Ruiz-Ordaz, N., J.C. Ruiz-Lagunez, J.H. Castanon-Gonzalez, E. Hernandez-Manzano, E. Cristiani-Urbina and J. Galindez-Mayer, 2001. Phenol biodegradation using a repeated batch culture of Candida tropicalis in a multistage bubble column. Revista Latinoamericana de Microbiologia, 43: 19-25.
Direct Link - Chang, Y.H., C.T. Li, M.C. Chang and W.K. Shieh, 1998. Batch phenol degradation by Candida tropicalis and its Fusant. Biotechnol. Bioengin., 60: 391-395.
Direct Link - Hubble, B.R., J.R. Stetter, E. Gebert, J.B.L. Harkness and R.D. Flotard, 1981. Experimental measurements from residential wood-burning stoves. Proceedings of the International Conference on Residential Solid Fuels: Environmental Impacts and Solutions, Jun. 1-4, Portland Hilton, Portland, Oregon, USA., pp: 79-138.
- Lowry, O.H., N.J. Rosebrough, A.L. Farr and R.J. Randall, 1951. Protein measurement with the folin phenol reagent. J. Biol. Chem., 193: 265-275.
CrossRefPubMedDirect Link