
Gholamreza Zarrini
Department of Bacteriology,
Zargham Sepehrizadeh
Department of Pharmaceutical Biotechnology, Faculty of Pharmacy,
Tehran University of Medical Sciences
Mojtaba Tabatabaei Yazdi
Department of Pharmaceutical Biotechnology, Faculty of Pharmacy,
Tehran University of Medical Sciences
Qorbane Behzadiyan Nejad
Department of Bacteriology,
Zuhair Muhammad Hassan
Department of Immunology, Faculty of Medical Sciences,
Tarbiat Modares University, Tehran, Iran
Pakistan Journal of Biological Sciences
Year: 2006 | Volume: 9 | Issue: 6 | Page No.: 1128-1131







ABSTRACT
Brucella abortus ribosomal L7/L12 protein is a 12 kDa protein encoded by rplL. This gene was amplified by PCR from B. abortus S19 and cloned in pET16b expression vector. The construct was transformed in Escherichia coli BL21 (DE3) PlysS. The expression was performed by addition 1 mM IPTG (isopropyl-beta-D-thiogalacto- pyranoside) in the medium with OD = 0.6 and incubation continued for 1, 2, 4 and 8 h at 37°C. The highest production (23 mg L-1) level was achieved after 4 h incubation of induction. The recombinant protein was purified by Ni-NTA column and characterized by SDS-PAGE.
PDF Abstract XML References Citation
How to cite this article
Gholamreza Zarrini, Zargham Sepehrizadeh, Mojtaba Tabatabaei Yazdi, Qorbane Behzadiyan Nejad and Zuhair Muhammad Hassan, 2006. Cloning and Overexpression of rplL Gene of Brucella abortus in Escherichia coli. Pakistan Journal of Biological Sciences, 9: 1128-1131.
DOI: 10.3923/pjbs.2006.1128.1131
URL: https://scialert.net/abstract/?doi=pjbs.2006.1128.1131
DOI: 10.3923/pjbs.2006.1128.1131
URL: https://scialert.net/abstract/?doi=pjbs.2006.1128.1131
INTRODUCTION
Brucellosis is a disease affecting various domestic and wild life species resulting from infection with bacteria belonging to the genus Brucella. It also causes undulant fever, endocarditis, arthritis and Osteomyelitis in humans (Young; 1983; Corbel, 1997; Ko and Splitter, 2003) Vaccination against brucellosis in cattle relies on live attenuated strains such as B. abortus strains S19 or RB51 (Ko and Splitter, 2003). Although these vaccines are effective in promoting a protective immune response, B. abortus strain 19 is pathogenic to humans and pregnant cattle (Young; 1983; Ko and Splitter, 2003). Furthermore there are no human vaccines currently available. Alternative vaccination strategies have recently been investigated. An antigen of B. abortus that elicits a cell-mediated immune response and confers protective immunity in mice is the ribosomal protein L7/L12 (Oliveira and Splitter, 1996; Kurar and Splitter, 1997). Brucella ribosomal L7/L12 proteins are 12 KD proteins encoded by rplL gene. L7 and L12 differ from each other only by an acetylic posttranslational modification that occurs at the L12 N-terminal and converting it to L7 (Bachrach et al., 1994, 1997).
Ribosomal proteins of intracellular pathogens were shown to be able to induce delayed type hypersensitivity (DTH) reaction in animal sensitized with the pathogen (Tantimavanich et al., 1993; Bachrach et al., 1994). Oliveira and colleagues produced Brucella L7/L12 as a fusion form with maltose binding protein (MBP). Bachrach et al., (1997) also produced histidine tagged form of Brucella ribosomal protein and demonstrated that tag did not interfere with the DTH activity. They produced 8 mg L–1 of E. coli culture medium.
In this study, in order to produce high level of Brucella ribosomal L7/L12 protein we cloned the rplL gene into a pET expression vector and expression was studied.
MATERIALS AND METHODS
Bacterial strains and growth conditions: B. abortus S19 (obtained from Razi Institute) was grown in Brain Heart Infusion (Difco) at 37°C for 48 h. E. coli BL21 (DE3) and E. coli BL21 (DE3) PlysS (Novagen) were cultured on Luria-Bertani medium at 37°C. Transformed E. coli colonies were selected on LB medium with 50 μg μL–1 ampicillin.
Cloning of B. abortus rplL gene in pET16b: The primers L7F (GAATTCCATATGGCTGATCTCGCAAAGA TCGTT G) and L7R (ACTAGTCTCGAGCTTGAGTTC AA CCTTGGCGC) were selected based on the B. abortus rplL gene sequence in Genbank (accession No. AF-169147). The primers included a NdeI cut site at the 5’end (L7F) of the gene and a XhoI site at the 3’ end (L7R). rplL gene was amplified from B. abortus by PCR. The PCR product was subjected to 1% Agarose gel and after primary confirm, it was purified with QIA quik® Gel extraction kit (Qiagen 28704). Then, the purified product was digested with NdeI and XhoI (Fermentas). The same digestion reaction was carried out on pET16b (Novagen). Ligation was done with T4 ligase (Roche). After ligation (overnight, at 10°C), the resulting plasmid containing rplL gene sequence, was used to transform E. coli BL21 (DE3). All reactions such as PCR, digestion, ligation and transformation procedures were preformed by standard literatures and manufacturer’s instructions.
Cloning confirmation: To confirm the presence of the recombinant plasmid in the transformed cells, the plasmid was extracted from the cells by High pure plasmid Isolation kit (Roche). The plasmid was digested by NdeI and XhoI. Then it was subjected to 1% Agarose gel and also, the recovered plasmid was further analyzed by PCR. The PCR was carried out with T7 primers and the PCR product was sent for sequencing (MWG, Germany).
Expression of recombinant L7/L12 protein: To assess the expression of L7/L12, the positive clones were cultured in LB medium containing 50 μg μL–1 ampicillin. Then, 1 mM isopropyl-beta-D-thiogalacto-pyranoside (IPTG) was added as an inducer to the medium with OD = 0.6 and incubation was continued for 1, 2, 4, 6 and 8 h. The cells were harvested and used for SDS-PAGE analysis. Gel was stained with coomassie blue and quantity of expressed protein was estimated by comparing of intensity of the protein bands.
Purification of L7/L12 protein: The cell mass which harvested from production medium was lysed and the recombinant L7/L12 protein was purified by Ni-NTA column as specified by the manufacture’s instructions (Qiagen cat No. 31314). The purified protein was dialyzed against saline and purify was analyzed by SDS-PAGE.
RESULTS
Construction of rplL expression vector: B. abortus rplL gene was amplified by PCR with the primers L7F and L7R. The resulting product had a size of 393 bp. It was according to the expected size of the gene (Fig. 1). For the insertion of the gene, recognition sites for NdeI and XhoI were introduced on the L7F and L7R, respectively. The same recognition sequences on the polylinker site of pET16b made the ligation reaction possible in correct direction.
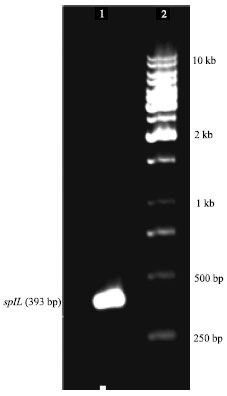 | |
| Fig. 1: | Amplification of rplL gene by PCR. Lane1: rplL gene PCR product; Lane 2: 1 kb DNA ladder |
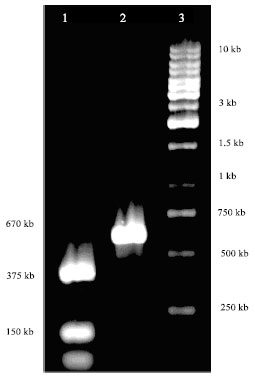 | |
| Fig. 2: | Digestion of PCR product was amplified by T7 primers. Lane1: The PCR product was digested by NdeI and XhoI and a band around 150 bp is a flanked DNA on both sides of the rplL gene; Lane2: The product of rplL gene was amplified by T7 primers; Lane3: 1 kb DNA ladder |
 | |
| Fig. 3: | rplL gene sequence, 375 bp |
 | |
| Fig. 4: | Expression of B. abortus L7/L12 protein in E. coli. LMW size marker; Lane1: PlysS E. coli cell lysate as a control, Lane 2: Expressed recombinant brucella L7/L12 protein after 4 h incubation |
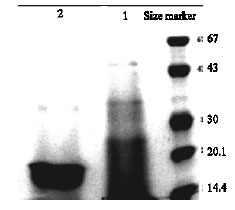 | |
| Fig. 5: | The recombinant L7/L12 was purified by Ni-NTA. Lane 1: Flow through of Ni-NTA column, Lane 2: The purified brucella ribosomal L7/L12 protein |
After the insertion of rplL, the construct was transformed in E. coli BL (DE3) plysS and the positive clones were selected. For the preliminary confirmation of the cloning, the plasmid was isolated from transformants and digested by NdeI and XhoI. In the positive transformants a 375 bp band was visible on the gel. On the other hand, A PCR reaction was performed with T7 primers on the vector, out of the insert sequence. The PCR product has a size around 650 bp. It was digested by NdeI and XhoI and 375 bp fragment was confirmed on the gel (Fig. 2).
The cloned rplL gene was sent to sequence (MWG, Germany). The sequence of the gene was the same with the sequences on the NCBI exception two nucleotides at G195C and C312G (Fig. 3).
Expression of B. abortus L7/L12 protein: The recombinant protein was expressed by addition of 1 mM IPTG as an inducer. The optimum incubation time after addition of IPTG was 4 h. After 4 h of incubation, around 23 mg of recombinant L7/L12 was produced in one liter of E. coli culture. The expressed protein was an intracellular protein and was detected only in cell lysate. SDS-PAGE analysis was shown a new band around 17.2 kDa (Fig. 4). This band matches with the expected molecular weight for recombinant B. abortus L7/L12 protein. It is a 12 kDa protein, but in the recombinant form as a result of fusion fragment of ten-histidine in the N-terminal. This tag was used for purification by Ni-NTA affinity column.
Purification of the recombinant protein: The production culture was prepared in 650 mL and the cell mass was harvested and subjected to purification procedure as specified by manufacturer’s instruction of Ni-NTA affinity column. The expected band was obtained in eluted fraction (Fig. 5).
DISCUSSION
The brucella L7/L12 protein is a candidate to design new vaccines against brucella infection and some efforts have been performed to use of this protein as a subunit and DNA vaccine. It seems that during infection by intracellular multiplying bacteria such as brucella, some of bacterial structural proteins, like the L7/L12 protein are involved in development of the cellular immune response (Bachrach et al., 1997; Ko and Splitter, 2003).
It was established that the L7/L12 is capable to inducing lymphocyte proliferation (Brooks-Worrell and Splitter, 1992; Oliveira et al., 1994). This protein has also showed DTH reaction in the sensitized guinea pig. It is present in Brucellergen that was used to DTH skin test (Bachrach et al., 1994).
pET16b is a vector that permits high expression and rapid purification of the recombinant protein through a His-tag fused to the expressed protein at N-terminus and in most cases the concomitant accumulation of the desired protein in high concentration (40-50% of the total cell protein). For the Brucella L7/L12 protein, production was achieved to 23 mg L–1 of E. coli culture after 4 h of addition IPTG. It was more than amount has reported before (Bachrach et al., 1994).
High level production of recombinant proteins facilate production of large amount of the desired protein, at the same time the limitation of overexpression of the heterologous proteins should be considered. They are often unable to obtain a native conformation or completely segregated within inclusion bodies. This problem may not be important for some antigenic proteins such as L7/L12 and these types of proteins can be produced in this system as a vaccine candidate successfully.
REFERENCES
- Bachrach, G., M. Banai, Y. Fishman and H. Bercovier, 1997. Delayed type hypersensitivity activity of the Brucella L7/L12 ribosomal protein depends on posttranslational modification. Infect. Immun., 65: 267-271.
Direct Link - Bachrach, G., D.B. Nir, M. Banai and H. Bercovier, 1994. Identification and nucleotide sequence of Brucella melitansis L7/L12 ribosomal protein. FEMS Microbial. Lett., 120: 237-240.
CrossRef - Brooks-Worrell, B.M. and G.A. Splitter, 1992. Antigens of Brucella abortus S19 immunodominant for bovine lymphocytes as identified by one- and two-dimensional cellular immunoblotting. Infect. Immun., 60: 2459-2464.
PubMedDirect Link - Ko, J. and G.A. Splitter, 2003. Molecular host pathogen interaction in brucellosis current understanding and future approaches to vaccine development for mice and humans. Clin. Microbiol. Rev., 16: 65-78.
CrossRef - Oleinikov, A.V., B. Peroud, B. Wang and R.R. Traut, 1993. Structural and functional domains of Escherichia coli ribosomal protein L7/L12. J. Biol. Chem., 268: 917-922.
Direct Link - Oliveira, S.C. and G.A. Splitte, 1994. Subcloning and expression of the Brucella abortus L7/L12 ribosomal gene and Y lymphocyte recognition of the recombinant protein. Infect. Immun., 62: 5201-5204.
Direct Link - Oliveira, S.C. and G.A. Splitter, 1996. Immunization of mice with recombinant L7/L12 ribosomal protein confers protection against Brucella abortus infection. Vaccine, 14: 959-962.
CrossRef - Ribeiro, L.A., V. Azevedo, Y.L. Loir, S.C. Oliveira and P. Langella et al., 2002. Production ad targeting of Brucella abortus antigen L7/L12 in lactococcus lactis a first step towards food grade live vaccines against brucellosis. Applied Environ. Microbiol., 68: 910-916.
Direct Link - Rice, P.A. and T.A. Steitz, 1989. Ribosomal protein L7/L12 has a helix turn helix motif similar to that found in DNA binding regulatory proteins. Nucleic Acids Res., 17: 3757-3762.
Direct Link - Tantimavanich, S., S. Nagai, H. Nomaguchi, M. Kinomoto, N. Ohara and T. Yamada, 1993. Immunological properties of ribosomal proteins from Mycobacterium bovis BCG. Infect. Immun., 61: 4005-4007.
Direct Link