
A. Ansari
Present Address: Department of Genetics, University of Karachi, Karachi, Pakistan
N. L. McQueen
Department of Biology/Microbiology, California State University, Los Angeles, California, USA
Pakistan Journal of Biological Sciences
Year: 1999 | Volume: 2 | Issue: 4 | Page No.: 1283-1285







ABSTRACT
We report here on the expression of the hemagglutinin- neuraminidase protein of the Sendai virus using a T7 RNA polymerase system in BHK-21 cell line. The expression was confirmed by the immunoprecipitation and Western/lmmunoblott techniques. This HN gene expression has confirmed that the T7 RNA polymerase and recombinant vaccinia virus systems can be used for the expression studies. In future we will try to express the HN gene along with other Sendai viral genes in the polarized cells and will try to locate the transport signals in their polypeptide backbones.
PDF Abstract XML References Citation
How to cite this article
A. Ansari and N. L. McQueen, 1999. Expression of Sendai Virus HN Gene in BHK-21 bell Line. Pakistan Journal of Biological Sciences, 2: 1283-1285.
DOI: 10.3923/pjbs.1999.1283.1285
URL: https://scialert.net/abstract/?doi=pjbs.1999.1283.1285
DOI: 10.3923/pjbs.1999.1283.1285
URL: https://scialert.net/abstract/?doi=pjbs.1999.1283.1285
INTRODUCTION
Enveloped RNA viruses when infect polarized cells bud ether from apical domain or the basolateral domain. Studies indicate that their envelope glycoproteins are found in the same domain from which the virus ultimately buds (Boulan and Pendergast, 1980). This suggests that these proteins contain sorting signals in their polypeptide backbones that direct their polarized transport. Viral proteins utilize the same transport and sorting machinery as endogenously synthesized proteins. Thus envelope RNA viruses have been extensively used as a model for the study of membrane protein transport. We report here on the transient expression of the Sendai HN protein using a T7 RNA polymerase system in nonpolarized BHK-21 cells and later it will be expressed in the polarized cells.
MATERIALS AND METHODS
Bacteria, viruses, plasmid and cells: E. coil strain MV1190 Wild type Sendai virus (Z strain) and the F1-R variant of Sendai were provided by Dr. Seto, California State University Los Angeles. Baby Hamster kidney cells (BHK-21) used for the expression of the HN gene was provided by Dr. Nayak. University of California Los Angeles and were grown as described earlier (McQueen et al., 1984).
Recombinant KS+ plasmid having T7 and T3 polymerase promoters flanking the polylinker region used for transcribing HN gene and vTF7-3 a recombinant vaccinia virus that expresses T7 polymerase that binds to T7 promoters to begin transcription were provided by Dr. Nayak, UCLA, Sendai HN cDNA with EcoR1 linkers was given by Elizabeth Cal. State LA.
Antibodies: Polyclonal anti Sendai rabbit IgG (primary antibody) and alkaline phosphatase-conjugated goat affinity purified anti rabbit IgG (secondary antibody) were given by Dr. Nayak, UCLA.
Cloning of HN gene to KS+ plasmid and transformation in MV1190 cells: After cloning HN genes in KS+ plasmid at EcoR1 site, MV1190 competent cells were used for transformation to produce the clone on a large scale and confirmed for the correct orientation of the insert and the concentration of the DNA by digesting with the Pst1 enzyme and electrophoresis on a 1 percent agarose gel stained with ethidium bromide.
Infection and transfection for HN gene expression: BHK-21 cells were grown in 60 cm2 tissue culture plates to 80 percent confluency as described earlier (McQueen et al., 1984). Recombinant vaccinia virus (vTF 7-3) and wt Sendai virus(for +ve control) were trypsinized with equal volume of 0.25 mg/ml trypsin for 30 minutes. One plate was infected with Sendai virus (1 pfu/cell) and two plates were infected with recombinant vaccinia virus (1 pfu/cell). The viruses were allowed to adsorb. for 1 hour at 37°C with rocking after every 15 minutes. During the last 15 minutes recombinant plasmid DNA was precipitated with lipofectin (15 μg of plasmid DNA in 70 μl volume was added to 30 μl lipofectin (1 unit/μl) in a polystyrene tube followed by a 15 minutes incubation at room temperature. This was then used for transfecting the cells. The transfected cells were incubated for 5 hours with serum free DMEM at 37°C in a humidified CO2 incubator followed by addition of 3 ml DMEM containing 20 percent FCS and 12 hours incubation.
Expression studies
lmmunoprecipitation: lmmunoprecipitation was done as described by McQueen et al. (1987). After the infection /transfection the cells were washed twice with 1X PBS (8 g NaCI, 1.15 g Na2HPO4, 0.2 g KCI, 0.2 g KH2PO4 followed by addition of 1 ml methionine free media. The cells were incubated for 1 hour after which labeling was done with 50 me of S35 methionine in 1 ml of methionine free media for 5 hours at 37°C in a humidified CO2 incubator. Labeled cells were lysed and immunoprecipitated with 1 ml of anti Sendai monoclonal IgG 1:3 diluted in RIPA buffer (10 mM tris, 1 percent deoxycholic acid, 1 percent tritonX-100, 1M phenyl methyl sulfonyl fluoride, 150 mM NaCI). One half of each immunoprecipitate was then analyzed by 10 percent SDS-PAGE. After wards the gel was dried and exposed to X-ray film and was developed after one week.
Western/lmmunoblot: After the infection/transfection of BHK-21 cells, as described earlier, the cells were first washed twice with 3 ml of cold PBS and scrapped twice in 750 ml of PBS. The suspension was centrifuged for 2 minutes at 4°C then to the pellet 200 ml of RIPA + Kallikrein inhibitor was added followed by incubation on ice for 10 minutes. Centrifugation was again performed at 4°C and the supernatant was saved at -20°C till loading on 10 percent protein gel, after the addition of 60 ml sample buffer. The running conditions were 650V, 15 m amp, 250 Watts. Next the protein from the gel was transferred to the nitrocellulose paper with the help of the electric blotter at 25 Volts, 450 m amp current and 250 watts for about 30 to 45 minutes. The paper was then stained for 5 minutes in 10 percent Ponceau S solution to check the transfer. To prevent the non specific binding of the antibody, the paper was blocked in the blocking buffer (1X PBS with MgCl2 and CaCl2, 0.005 percent tween 20 and 3 percent non fat dry milk). After this polyclonal anti Sendai rabbit IgG (primary antibody) was added in 1:1000 blocking buffer) so that it could bind to the protein. Washing with the blocking buffer was done to remove the nonspecifically bounded antibodies. The specifically bound antibodies were detected by the addition of alkaline phosphatase-conjugated goat affinity purified anti rabbit lgG (secondary antibodies) with the concentration of 1:1000 in blocking buffer. After one hour incubation at room temperature antibody bound proteins were visualized by staining solution {10 ml of 100 mM tris HCI, pH 9.5+32 μl BCIP (5-bromo-4-chloro-3-indolyl phosphate)} as a substrate for alkaline phosphate and 66 oul NBT (p-nitro blue tetrazodium chloride) as a color developer.
RESULTS
Cloning of HN gene to KS+ plasmid: The HN gene was ligated to KS+ plasmid predigested with EcoR1. KS+ containing the ligated HN gene was transformed into MV1190 cells made competent with CaCl2. Twelve white recombinant clones were selected randomly for miniscreening for the correct orientation of the gene for the use of Vaccinia/T7 expression system. Bands of 3386, 360, 264 and 486 base pairs were obtained when the HN gene was in the correct orientation and bands of 3386, 264, 360 and 976 base pairs were obtained when the HN gene was in the wrong orientation. Out of 12 clones screened, 4 were found to have HN gene in correct orientation. One clone was selected randomly for the future use.
Expression studies
Immunoprecipitation: Newly synthesized HN protein was labeled with S35 methionine after 1 hour starvation for methionine. The cells immediately utilized the methionine for the synthesis of HN protein. This was confirmed by exposing a Fuji RX, N1F X-ray film with an intensifier to the protein gel. Sendai virus infected cells were used as a positive control. The HN protein band was found at the same position on the gel as the that of the HN control (Fig. 1).
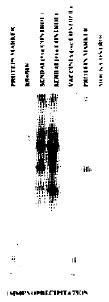 | |
| Fig. 1: | Image of X-ray film developed after the exposure to 10 percent SOS polyacrylamide gel. The band of protein in KS+/HN is at the same position as in the case of Sendai virus HN positive control. Vaccinia Virus negative control and mock control shows no band at that position |
 | |
| Fig. 2: | Image of the nitrocellulose paper on which the samples from the protein gel were transfered a were treated with polyclonal anti Sendai rabbit lgG and alkaline phosphatase-conjugated goat affinity purified anti rabbit IgG. Finally it was treated with BCIP and NBT to visualize the results. The band of KS+/HN was found at the same position as the HN of different Sendai virus strains. BHK cells and Vaccinia vTF negative controls showed no bands |
Western/Immunoblot: The cells were allowed to synthesize the HN protein and the protein was then allowed to bind with the primary antibody (anti-Sendai rabbit lgG), followed by a secondary antibody (alkaline phosphatase conjugated goat affinity anti-rabbit IgG). The complex was stained as described earlier and photographed. Sendai Virus protein was used as a positive control. The sample showed a band running at the same distance from the wells as the Sendai HN control. Thus the mobility of protein in the gel and its reactivity with the antiserum suggests that the HN protein is synthesized and glycosylated (Fig. 2).
DISCUSSION
Sendai virus is a negative stranded RNA virus that causes the pulmonary diseases in mice. Six structural proteins of the virus have been identified of which HN is a multifunctional molecule that is responsible for binding of the virus to the sialic acid containing receptors on host cells, agglutination of erythrocytes, neuraminidase activity and antigenic properties. It is also believed to play a possible role in fusion (Mostov et al., 1992). The precise locations of the Hemagglutinin and neuraminidase activities have not been identified.7The HN gene was ligated into KS+ vector, at the EcoR1 site, followed by transformation into MV1 190 competent cells. KS+ plasmid also known as Bluescript vector has two selectable markers which were used for the selection of appropriate clone. The first is the ampicillin resistance gene that helped in the selection of transformed cells via their ability to grow on L-ampicillin plates. The second is the (3-galactosidase gene that contained the polylinker region. This helped to screen clones having HN gene insert. After confirming the presence of the HN gene inserts in the clones, they were checked for the correct orientation for the use in the expression studies by digesting them with the Pst1 enzyme. Four out of twelve clones showed correct orientation of the HN gene. One of the clone was then selected randomly for the expression studies and its DNA was amplified by growing the organism on a large scale.
HN cDNA was inserted into the Bluescript vector downstream of the T7 RNA polymerase promoter. T7 RNA was provided by the recombinant vaccinia virus vTF7-3 (Fuerst et al., 1987). Infection of the BHK-21 cells with v77-3 was followed by the transfection with HN/KS+ DNA. The T7 RNA polymerase expressed by vTF7-3 bounded to the T7 promoter on KS+ present in the cytoplasm and the HN gene was transcribed and then translated. There were several reasons for using the recombinant Vaccinia virus T7 polymerase system in our protein expression studies. T7 RNA polymerase is highly selective for its own promoters. A relatively small amount of T7 RNA polymerase is sufficient to direct high level transcription from a T7 promoter in a multicopy plasmid. T7 RNA polymerase appears to be capable of transcribing any DNA linked to a T7 promoter so the T7 expression should he capable of transcribing any gene (Studier and Moffatt, 1986).
Transfection was done using lipofectin reagent which was found to give higher degree of transfection efficiency as compared to conventional calcium phosphate method (Felgner et al., 1987).
Two different methods, immunoprecipitation and immuno/Western blotting were used to detect expression of the HN protein . They have been successfully used in several previous studies (McQueen et al., 1987). The HN protein produced by the transient expression system was compared with positive (Sendai Virus Infection) and negative (Vaccinia Virus Infection) controls. The expressed HN protein showed the same migration rate as the HN protein produced from the Sendai virus infected cells. These results indicated that HN proteins were synthesized and properly glycosylated in the BHK-21 cells.
ACKNOWLEDGEMENT
Support for the research was provided in part by the National Institute of health, US. Public Health Service grants Al 311191-01 and 5 806 GM08101-26 to N.L. McQueen.
REFERENCES
- McQueen, N.L., D.P. Nayak, L.V. Jones and R.W. Compans, 1984. Chimeric influenza virus hemagglutinin containing either the NH2 terminus or the COOH terminus of G protein of vesicular stomatitis virus is defective in transport to the cell surface. Proc. Nat. Acad. Sci., 81: 395-399.
Direct Link - McQueen, N.L., D.P. Nayak, E.B. Stephens and R.W. Compans, 1987. Basolateral expression of a chimeric protein in which the transmembrane and cytoplasmic domains of vesicular stomatitis virus G protein have been replaced by those of the influenza virus hemagglutinin. J. Biol. Chem., 262: 16233-16240.
Direct Link - Mostov, K., G. Apodaca, B. Aroeti and C. Okamoto, 1992. Plasma membrane protein sorting in polarized epithelial cells. J. Cell Biol., 116: 577-583.
Direct Link - Felgner, P.L., T.R. Gadek, M. Holm, R. Roman and H.W. Chan et al., 1987. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Nat. Acad. Sci., 84: 7413-7417.
Direct Link - Boulan, E.R. and M. Pendergast, 1980. Polarized distribution of viral envelope proteins in the plasma membrane of infected epithelial cells. Cell, 20: 45-54.
CrossRefDirect Link - Studier, F.W. and B.A. Moffatt, 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol., 189: 113-130.
CrossRefPubMedDirect Link - Fuerst, T.R., P.L. Earl and B. Moss, 1987. Use of a hybrid vaccinia virus-T7 RNA polymerase system for expression of target genes. Mol. Cell. Biol., 7: 2538-2544.
CrossRefDirect Link