
M.C. Sharma
School of Pharmacy, Devi Ahilya Vishwavidyalaya, Indore (M.P) 452001, India
Smita Sharma
Department of Chemistry, Chodhary Dilip Singh Kanya Mahavidyalya Bhind (M.P)-477001, India
Journal of Pharmacology and Toxicology
Year: 2011 | Volume: 6 | Issue: 5 | Page No.: 493-504







ABSTRACT
Antitubercular activity of 7-methyljuglone derivatives series were subjected to quantitative structure activity relationship analysis with an attempt to derive and understand a correlation between the biological activity as dependent variable and various descriptors as independent variables. Several statistical regression expressions were obtained using multiple linear regression analysis. The QSAR models were generated using 19 compounds. The predictive ability of the resulting QSAR models was evaluated employing the leave one-out method of cross validation. Several statistical regression expressions were obtained using multiple linear regression analysis. The analysis of best resulted in the following 2-D model which suggests that pIC50 = [-0.025] MR+[0.278] StrE+[0.028] p+[3.04459] HOMO, n = 19, r = 0.87961, r2 = 0.81048, variance = 0.0805, SD = 0.4324, F = 85.78. The study suggested that substitution of group at R1 and R3 position on naphthoquinones ring with hydrophobic nature and low bulkiness are favorable for the antitubercular activity in the concerned microbes. The quantitative structure activity relationship study provides important structural insights in designing of potent antitubercular agents.
PDF Abstract XML References Citation
Received: December 07, 2010;
Accepted: March 27, 2011;
Published: May 31, 2011
How to cite this article
M.C. Sharma and Smita Sharma, 2011. 2D Qsar Study of 7-Methyljuglone Derivatives: An Approach to Design Anti Tubercular Agents. Journal of Pharmacology and Toxicology, 6: 493-504.
DOI: 10.3923/jpt.2011.493.504
URL: https://scialert.net/abstract/?doi=jpt.2011.493.504
DOI: 10.3923/jpt.2011.493.504
URL: https://scialert.net/abstract/?doi=jpt.2011.493.504
INTRODUCTION
The emergence of drug resistant strains of Mycobacterium tuberculosis, particularly multiple drug resistant strains has complicated treatment protocols and raises the concern that tuberculosis may once again become an incurable disease. For this reason it is critical to discover new drugs acting with a mechanism different from those of presently used antitubercular drugs. Tuberculosis (TB), caused by M. tuberculosis, is a major public health and socioeconomically problem in most of the developing countries (Heym and Cole, 1997; Basso and Blanchard, 1998; Telenti and Iseman, 2000). Tuberculosis (TB) is a contagious disease. Like the common cold, it spreads through the air. Only people who are sick with TB in their lungs are infectious. When infectious people cough, sneeze, talk or spit, they propel TB germs, known as bacilli, into the air. A person needs only to inhale a small number of these to be infected. Left untreated, each person with active TB disease will infect on average between 10 and 15 people every year. Despite the ready availability of effective treatments, tuberculosis remains a major public health threat worldwide. With approximately one-third of the world population currently infected, increased prevalence of the disease in HIV-infected patients and the emergence of multi-drug-resistant bacteria, TB remains a major world health problem. Hence there is a continuing need to find additional lead compounds and biological targets for novel antitubercular chemotherapies. The acid-fast bacillus Mycobacterium tuberculosis is the causative agent of Tuberculosis (TB). The tubercle bacillus is a slow growing organism, which does not elicit a sharp and massive reaction from the host. The tubercle bacillus does not produce any substance, which is toxic to the normal host. It acts as an irritating foreign body and tubercle formation can be produced by virulent, avirulent and nonpathogenic types. The tubercle bacillus is an intracellular parasite and lives and grows within the host’s tissue cells, macrophages and epithelial cell. The efficacy of the currently available agents used in standard Tuberculosis (TB) treatment regimens is severely limited by several factors; including long treatment regimens, multiple drug treatment regimens, drug interactions and drug resistance (WHO, 1995). Drug resistance and multidrug-resistant tuberculosis is perceived as a growing hazard to human health worldwide. TB ranks among the most important burdens on human health, not only due to the large number of cases (~9 million/year worldwide, with incidence rates typically measured per 100,000 population), but also because about one quarter of sufferers die, most of them young adults. Globally, the number of TB cases is currently rising at 2% year-1. The fear is that the number of cases resistant to antitubercular drugs may be increasing much faster (Dye et al., 2002; Dye and Williams, 2000; Pablos-Mendez et al., 2002; Petrini and Hoffner, 1999). The major concerns over drug resistance were a fear of the spread of drug-resistant organisms and the ineffectiveness of chemotherapy in patients infected with them. If these spread increasingly in a community, TB may become progressively uncontrollable using currently available chemotherapy. One of the strategies suggested for overcoming this problem is the fully exploiting the potential of standard short course chemotherapy based on cheap and safe first line drugs. Furthermore, the development of potent new antitubercular drugs without cross-resistance with known antimycobacterial agents is urgently needed (Tomioka, 2002). According to statistics, one-third of the world’s population is currently infected with the TB bacillus, each year, 8 million people world-wide develop active TB and about 1.7 million people die (http://www.who.int/tb/en/.). Currently, an important problem in TB treatment is the development of multi-drug resistant tuberculosis (MDR-TB), which can be defined as strains that are resistance to at least isoniazid and rifampicin, important first line drugs used in TB treatment. Therefore, there is a need for new drugs of new structural classes and with a novel mechanism of action other than isoniazid (INH), Rifampicin (RIF) and Pyridazinamide (PZA). In this regard, since the last decade search for new antitubercular substances has ranked among the priority areas of chemotherapeutic research. The spread of MDR-TB could cost between 100 and 1400 times the available treatment costs and further threatens to make TB incurable. Exact data are hard to estimate but at least 4% of all world-wide TB patients are resistant to at least one of the current first line drugs. Another serious problem, in the context of MDR-TB, is the XDR-TB, abbreviation for extensively drug-resistant tuberculosis (TB) which are strains resistant to first and second line anti-TB drugs (De Souza 2006a, b). The dramatic increase in TB cases observed in the recent years is a result of two major factors. First is the increased susceptibility of people infected with Acquired Immunodeficiency Syndrome (AIDS) to TB, which augments the risk of developing the disease 100-fold with some showing cross-resistance to as many as nine drugs (El Sayed et al., 2000). A Quantitative Structure-activity Relationship (QSAR) enables the investigators to establish a reliable quantitative structure-activity and structure-property relationships to derive an in silico QSAR model to predict the activity of novel molecules prior to their synthesis. The overall process of QSAR model development can be divided into three stages namely, the data preparation, data analysis, and model validation, representing a standard practice of any QSAR modeling. The purpose of QSAR study is to find a relation between the composition or structure of a compound with its bio or chemical activity, in order to design a new compound with expected properties or predict the properties of an unknown compound (Gupta and Kumaran, 2006; Karthikeyan et al., 2006, 2007; Moorthy and Trivedi, 2006; Chaudhary et al., 2008; Gokhale and Kulkarni, 2000). A wide range of descriptors has been used in QSAR modeling. These descriptors have been classified into different categories, including constitutional, geometrical, topological, quantum chemical and so on. There are several variable selection methods including Multiple Linear Regression (MLR). To gain insight into the structural and molecular requirement influencing the antitubercular activities, we herein describe the QSAR analysis of 7-methyljuglone derivatives. The relevance of the model for the design of novel derivatives should be assessed not only in terms of predictivity, but also in terms of their ability to provide a chemical and structural explanation of their binding interaction. In this research, an attempt has been made to describe the Quantitative Structure-activity Relationship (QSAR) analysis of 7-methyljuglone derivatives to study and deduce a correlation between structure and antitubercular activity of these derivatives.
MATERIALS AND METHODS
Data set: The activity data of 7-methyljuglone derivatives was taken from the reported study of Mahapatra et al. (2007). The activity data have been given as IC50 values, where IC50 refers to the experimentally determined molar concentration of the 7-methyljuglone derivatives required to fifty percentage inhibitory concentrations. The biological activity values [IC50 (μg mL-1)] reported in the study were converted to nanomolar units and then further to-log scale and subsequently used as the response variable for the QSAR analysis. The -log values of IC50 along with the structure of compounds in the series is presented in Table 1. Initially the series was subjected to analysis (Fujita and Ban, 1971) using a regression technique to estimate the de novo contribution of substituents to the activity of the molecules. The method used in this study is a modification of the Free-Wilson technique. Here, the log of activity is considered to be a free energy related parameter which is additive in nature. Fujita and Ban derived a linear equation for a set of substituent’s, in the form of Eq. 1 as follows:
| (1) |
where, BA is biological activity, Gi is the log activity contribution or the log activity enhancement factor of the ith substituent relative to that of H and Xi is a parameter which takes a value of 1 or 0 according to the presence or absence of the ith substituent and μ = log BA, calculated for the unsubstituted compound, i.e., parent compound. The data was transferred to a statistical program in order to establish a correlation between physicochemical parameters as an independent variable and the antitubercular activity as a dependent variable using a sequential multiple linear regression analysis method (in sequential multiple regression the program searched for all permutation and combination sequentially for the data set).
Geometry optimization: The series was further subjected to molecular modeling studies using CS Chem- Office Software version 7.01 (Cambridge soft) (CS Chem Office, 2002). The structure of the compounds was drawn in Chem Draw Ultra version 7.01 and then copied to Chem 3D Ultra to create the three-dimensional (3D) model, which was saved as the template model. For every compound, the template model was suitably modified considering its structural features so that every compound maintained the same sequence of atoms.
| Table 1: | Structure and observed biological activity of series |
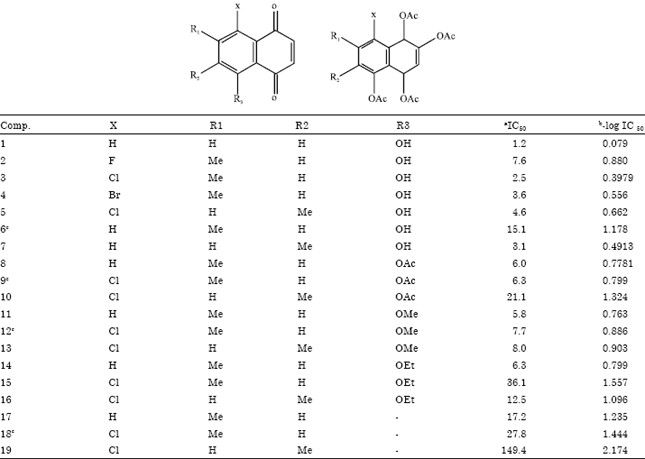 | |
| a:IC50 (in nM) was the in vitro observed biological activity of compounds, b: Negative logarithmic value of IC50, c: Test compounds | |
The molecular structures of all 19 compounds were sketched using the Chemdraw Ultra (Version, 7.01) software and energy minimized via MOPAC with energy tolerance value of root mean square gradient 0.001 kcal mol-1 and maximum number of iteration set to 1000. These structures were then subjected to energy minimization using force field molecular mechanics-2 (MM2) until the Root Mean Square (RMS) gradient value became smaller than 0.1 kcal mol-1. Å. Minimized molecules were subjected to re-optimization via Austin model-1 (Kier, 1971) method until the RMS gradient attained a value smaller than 0.0001 kcal mol-1 Å using MOPAC. The geometry optimization of the lowest energy structure was carried out using Eigenvector following routine. The descriptor values for all the molecules were calculated using compute properties module of programme. The energy minimized geometry was used for the calculation of physicochemical descriptor and extended Huckel charges of different atoms. These compound properties are the physicochemical descriptors that may be used to estimate the SAR of molecules. All conformers generated for each structure were analyzed in conformational geometrics panels with great care, and the lowest energy conformation of each structure was selected and added to a molecular database to compute various physicochemical properties. The descriptor values used in the model generation are shown in the Table 2.
| Table 2: | Calculated values of independent variables |
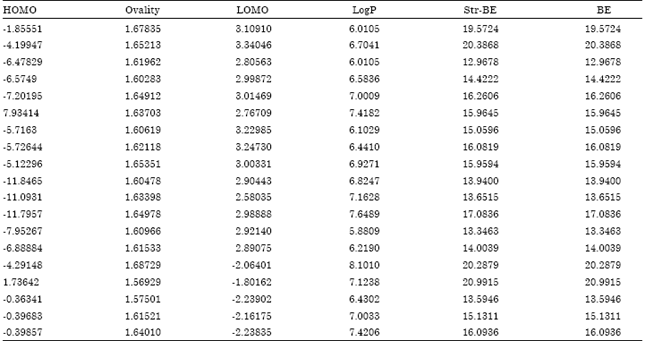 | |
Statistical methods and molecular descriptors: The values of substituent constants like hydrophobic (p), steric (Molar refractivity or MR), Hydrogen Acceptor (HA), Hydrogen Donor (HD) and electronic (field effect or F, resonance effect or R and Hammett's constant or s) were taken from the literature (Hansch and Leo, 1979). The series was divided in to a training set of 15 compounds and a test set of 4 compounds carried out automatically by the VALSTAT software. The sequential multiple linear regression analysis method was employed. In sequential multiple linear regression, the program searches for all permutations and combinations sequentially for the data set. The ± data with in the parentheses are the standard deviations associated with the coefficient of descriptors in regression equations. Calculated thermodynamic descriptors included Critical Temperature (Tc), Ideal Gas Thermal Capacity (Cp), Critical Pressure (Pc), Boiling Point (BP), Henry's Law Constant (H), Bend Energy (Eb), Heat of Formation (Hf), Total Energy (TE) and Partition Coefficient (PC). Steric descriptors derived were Connolly Accessible Area (CAA), Connolly Molecular Area (CMA), Connolly Solvent Excluded Volume (CSEV), Exact Mass (EM), Molecular Weight (MW), Principal Moment of Inertia-X component (PMI-X), Principal Moment of Inertia-Y Component (PMI-Y), Principal Moment of Inertia-Z Component (PMI-Z), Molar Refractivity (MR) and Ovality (OVAL). Electronic Descriptors Such as Dipole (DPL), Electronic Energy (ElcE), Highest Occupied Molecular Orbital Energy (HOMO), Lowest Unoccupied Molecular Orbital Energy (LUMO), Repulsion Energy (NRE), VDW-1,4-energy (E14), Non-1, 4-VDW energy (Ev) and total energy (E) were calculated. Stepwise multiple linear regression analysis method was used to perform QSAR analysis employing in-house VALSTAT (Gupta et al., 2004) programme establish a correlation between physicochemical descriptor used in this study as independent variable and Antitubercular activity as dependent variable using sequential multiple linear regression analysis method (in sequential multiple regression the program searched for all permutation and combination sequentially for the data set). The ±data within the parentheses are the error of regression coefficients associated with corresponding regression coefficients in regression equation. The best model was selected on the basis of various statistical parameters such as Correlation Coefficient (r), Standard Error of Estimation (SE), Sequential Fischer test (F) the Bootstrapping r2, chance, Q2 value, Spress value, Standard Deviation of Error Prediction (SDEP) and the predictive squared correlation coefficient of the test set (r2 pred) cross-validated squared correlation coefficient using leave one out procedure r2 chance statistics (evaluated as the ratio of the equivalent regression equations to the total number of randomized sets; a chance value of 0.001 corresponds to 0.1% chance of fortuitous correlation), outliers (on the basis of Z-score value) and predictive squared correlation coefficient of test set r2 pred r2. The squared correlation coefficient (or coefficient of multiple determination) r2 is a relative measure of fit by the regression equation. Correspondingly, it represents the part of the variation in the observed data that is explained by the regression. The correlation coefficient values closer to 1.0 represent the better fit of the regression. The F-test reflects the ratio of the variance explained by the model and the variance due to the error in the regression. High values of the F test indicate that the model is statistically significant. Standard deviation is measured by the error mean square, which expresses the variation of the residuals or the variation about the regression line. Thus standard deviation is an absolute measure of quality of fit and should have a low value for the regression to be significant. Quality of the each model was estimated from the cross-validated squared correlation coefficient (Kubinyi, 1993) calculated root mean square error (SDEP), chance statistics evaluated as the ratio of the equivalent regression equations to the total number of randomized sets; a chance value of 0.01 corresponds to 1% chance of fortuitous correlation and boot-strapping square correlation coefficient (r 2 subbs), which confirm the robustness and applicability of QSAR equation.
Multiple linear regression analysis: Multiple linear regression analysis and other statistical analysis were carried out on all the 19 molecules. The outlier molecules were then removed to improve the equation's predictive power. The final set of equations was obtained using 19 molecules and the best equation was obtained by using the optimal combination of descriptors. Descriptors were selected for the final equation based on their correlation coefficients and those descriptors having inter-correlation coefficient below 0.7 were considered, to select the best equation. Cross validation by leave one out method was carried out on these final set of 19 molecules to further enhance and validate the predictive power of the equation. Acceptability of the regression equation was judged by examining the statistical parameters. Correlation matrix was obtained to justify the use of more than one variable in the study. The variables used were with maximum correlation to activity and minimum inter-correlation with each other. From the statistical viewpoint, the ratio of the number of samples (N) to the number of variables used (M) should not be very low; usually it is recommended that N/M = 5. The QSAR equations were constructed for efficacy data of both species of malaria parasite with the physcio-chemical descriptors and indicator variables. The statistical quality of the equations was judged by the parameters like correlation coefficient (r), explained variance (r2), standard error of estimate (s) and the variance ratio or overall significance value (F). The accepted equations are validated for stability and predictive ability using leave-one-out and cross validation technique. The statistical parameters used to access the quality of the models are the Predictive Sum of Squares (PRESS) of validation. Finally, the standard cross-validation correlation coefficient r2 and q2 are also calculated:
PRESS = Σ (Ypred - Y obs)2 Spress = / PRESS/(n-k-1) |
Where:
| N | = | No. of compounds used for cross-validation |
| Yi | = | Experimental value of the physic-chemical property for the ith sample |
| Y | = | Value predicted by the model built without the sample i |
RESULTS AND DISCUSSION
When data set was subjected to stepwise multiple linear regression analysis, in order to develop 2D-QSAR between antitubercular activity in various microbes as dependent variables and substituent constants as independent variables, several models were obtained. Acceptability of the regression model was judged by examining the correlation coefficient (r), squared correlation coefficient (r2), F-test (F) and standard deviation. QSAR (Desai et al., 2001; Ghosh and Bagchi, 2010; Manvar et al., 2010; Narute et al., 2008; Sivakumar et al., 2007, 2010a, b; Sharma et al., 2010; Sharma and Sharma, 2010) in Biological activity data and various physicochemical parameters were taken as dependent and independent variables respectively and correlations were established using sequential multiple regression analysis in the help of design new molecules of antitubercular activity.
| • | pIC50 = [-0.025] MR +[0.278] StrE + [0.028] p + [3.04459] HOMO |
| • | n = 19, r = 0.87961, r2 = 0.81048, variance = 0.0805, SD = 0.4324, F = 85.78 |
Model-1 has a good correlation coefficient (r = 0. 87961), the model is tested for outlier by Z-score method and no compound was found to be an outlier, which suggested that the model is able to explain the structurally diverse analogs. The r2bs is 0.5964 as par with the conventional squared correlation coefficient (r2). Randomized biological activity test (p<0.014) revealed that the results were not based on chance correlation. The inter correlation among the parameters is 0.108. The cross validated squared correlation coefficient (Q2 = 0.7985), the predictive residual sum of square SPRESS = 0.2665) and the standard error of prediction (SDEP = 0.7259) suggested good internal consistency as well as predictive activity of the biological activity with high HOMO. The above model is validated by predicting the biological activities of the test molecules, as indicated in Table 3. The plot of observed versus predicted activities for the test compounds is represented in (Table 3). From Table 3 it is evident that the predicted activities of all the compounds in the test set are in good agreement with their corresponding experimental activities and optimal fit is obtained.
| • | pIC50 = [+1.158] + PMIY [-9.4057] +S-BE [0.156] +HOMO [0.024] +Ovality [0.365] |
| • | n = 15, r = 0.81627, r2 = 0.76205, SD. = 0.104, F = 7.636 |
Model-2 has a better correlation coefficient (r = 0.81627), the model is tested for outlier by Z-score method and no compound was found to be an outlier, which suggested that the model is able to explain the structurally diverse analogs. The inter correlation among the parameter of the descriptors is 0.108. The r2bs is 0.7513 as par with the conventional squared correlation coefficient (r2). Randomized biological activity test (p<0.001) revealed that the results were not based on chance correlation. The inter correlation among the parameters is 0.108. In general the model fulfills the statistical validation criteria to the significant extent however; it is a useful theoretical base for proposing more active compounds. In model PMIY,S-BE, HOMO contributed positively to the activity.
| Table 3: | Predicted biological activity and LOO predicted activity of QSAR models |
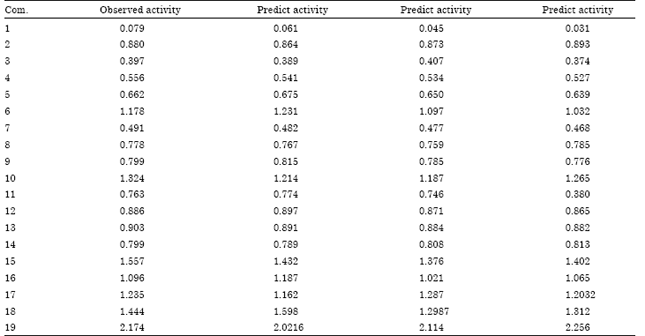 | |
The above model is validated by predicting the biological activities of the test molecules, as indicated in Table 3. The plot of observed versus predicted activities for the test compounds is represented in Table 3.
| • | pIC50 = 0.322 (±0.199) Log P+0.0237 (±0.019) DPL-10.189 ( ±0.341) LUMO |
| • | n = 15, r = 0.8038, r2 = 0.75321, variance = 0.00431, SD = 0.56396, F = 51.769 |
Model-3 fulfills many of the statistical validations such as the correlation coefficient; the cross validated squared correlation coefficient, standard deviation, bootstrapping squared correlation coefficient and chance. But the predictive residual sum of square standard error of prediction is less than 0.5 (0.35). The correlation accounted for more than 72.9% of the variance in the activity. The data showed an overall internal statistical significance level better than 99.9% as F (3, 16 α 0.001) = 51.769 which exceeds the tabulated F (3, 16 α 0.001) = 9.01, the cross validated squared correlation coefficient (Q2 = 0.7032), the predictive residual sum of square SPRESS = 0.1769) and the standard error of prediction (SDEP = 0.2784) suggested good internal consistency as well as predictive activity of the biological activity with high logP. In general the model fulfills the statistical validation criteria to the significant extent however; it is a useful theoretical base for proposing more active compounds. In model LUMO contributed negatively and LogP, DPL contributed positively to the activity. The above model is validated by predicting the biological activities of the test molecules, as indicated in Table 3 The plot of observed versus predicted activities for the test compounds is represented in Table 3.
| • | pIC50= [-2.392] +BE [-0.000108] +S-BE [0.212] +VDW [0.074] |
| • | n = 159, r = 0.74610, r2 = 0.72183, SD. = 0.123688, F = 45.3295 |
Model 4 (pIC50) has a better correlation coefficient (r = 0.74610), the model is tested for outlier by Z-score method and no compound was found to be an outlier, which suggested that the model is able to explain the structurally diverse analogs. The inter correlation among the parameter of the descriptors is 0.3967. The r2bs is 0.759 as par with the conventional squared correlation coefficient (r2). Randomized biological activity test (p<0.003) revealed that the results were not based on chance correlation. The inter correlation among the parameters is 0.108. In general the model fulfills the statistical validation criteria to the significant extent however; it is a useful theoretical base for proposing more active compounds. In model BE contributed negatively and S-BE, VDW contributed positively to the activity.
| • | pIC50= [-2.392] +BE [-0.000108] +MR [0.212] +VDW [0.074] + HOMO [0.243] |
| • | n = 19, r = 0.7309, r2 = 0.5338, SD. = 0.123688, F = 35.3295 |
Model-5 has a better correlation coefficient (r = 0.730), the model is tested for outlier by Z-score method and no compound was found to be an outlier, which suggested that the model is able to explain the structurally diverse analogs. The inter correlation among the parameter of the descriptors is 0.397. The r2bs is 0.759 as par with the conventional squared correlation coefficient (r2). Randomized biological activity test (p<0.003) revealed that the results were not based on chance correlation. The inter correlation among the parameters is 0.108. In general the model fulfils the statistical validation criteria to the significant extent however, it is a useful theoretical base for proposing more active compounds. In model BE contributed negatively and S-BE, VDW contributed positively to the activity.
Validation of the model: Model-1 shows a better correlation coefficient (r = 0.8796) which accounts for more than 87.96% of the variance in the activity, also the intercorrelation among the parameters is less (0.2315). The model shows that in the multi-variant model, the dependent variable can be predicted from a linear combination of the independent variables. The p-value is less than 0.001 for each physiochemical parameter involved in model generation. The data showed an overall internal statistical significance level better than 99.9% as it exceeded the tabulated F(3, 17 α 0.001) = 85.78. The model was further tested for the outlier by the Z-score method and no compound was found to be an outlier, which suggested that the model is able to explain the structurally diverse analog and is helpful in designing more potent compounds using physiochemical parameters. The leave-one-out cross validation method was employed for the prediction of activity (Table 3). The cross-validated squared correlation coefficient (in the biological activity data of leave-one-out) (Q2 = 0.7985), predictive residual sum of square (SPRESS = 0.2665), and standard error of prediction (SDEP = 0.7259) suggested a good internal consistency as well as predictive ability of the biological activity with low SDEP. The r2bs is at par with the conventional squared correlation coefficient (r2). Randomized biological activity results were not based on the correlation. The robustness and wide applicability of the model were further explained by significant r2pred value (0.81048) .In general, the model fulfils the statistical validation criteria to a significant extent to be a useful theoretical base for proposing more active compound. In model (1) p contributed positively where as Str-BE contributed positively towards biological activity is representative of atoms of hydrophobic nature in the molecules and suggests that substitution of groups, which are high hydrophobic in nature, might increase the biological activity. Thus, reimproving the p characteristics of the molecule increases the selective activity. Whereas minimizing the property like Str. BE which is helpful for rationalizing the interaction between molecule and receptor surface. The study revealed that distal end substitutions might interact with a hydrophobic pocket at receptor site, hence increasing hydrophobicity of the substituent increase the binding capacity between molecule and receptor surface which potentiate the selectivity as well as activity.
CONCLUSION
Although, generation of QSAR models with good statistical significance is of paramount importance, the models should also exhibit good predictive ability. 2D-QSAR analysis suggested that for all the, substitution at R1 position very much dominates the activity as compared to the indicator variable at R2 and R 3 position (Compound 1-16). At R1 position, logp contributed positively, which is responsible for hydrophobicity of the molecules; but MR contributed negative, which suggests that less bulky substitutions form the activity.
The series was also subjected to molecular modelling using 3D-QSAR; all the descriptor values for the molecules, calculated from the programme, were considered as independent variables. The 2D analysis suggested that the substitution at R1 position with various alkyl groups affect the antitubercular activity of naphthoquinones ring analogues as compared to substitutions at R 2 and R 3 position. The QSAR studies revealed that spatial parameter PMIY plays a significant role in explaining antitubercular activity of 7-methyljuglone. It contributed negatively to the expression, which suggested that less bulky groups around Y-axis in the molecules are favorable for the activity. DPL contributed positively to the activity up to a small extent as compared to the PMIY, suggesting that the moiety, which increases the charge distribution over the molecules, is favourable for the activity and optimising the hydrophobicity and bulkiness at R1 position. The developed QSAR model can be utilized for the further development of new molecules belonging to the class of 7-methyljuglone to exhibit good antitubercular activity, as it reveals the various physico-chemical parameters that play important roles in exhibiting potential antitubercular activity. The predictive ability of the models was gauged by a cross validation procedure following a leave-one-out scheme. All the models exhibit high q2 and low r2-se and q2-se values confirm their excellent predictive potential. The linear QSAR models have been successfully established using the different chemo metric tools like enhanced replacement method, multiple regressions. Predictive quality of the models was tested using predictive r2 (pred-r2 ), followed by a modified r2 (r2m) value based on training set, test set. (LOO) values for whole data set and training/test set are in agreement reflecting external validation characteristic of the developed QSAR models. The values of these parameters ensure the predictability, reliability and acceptability of the model. Based on all the statistical and validation parameters enhanced replacement method was found to give better results. QSAR models were proposed for antibacterial activity of the 7-methyljuglone derivatives using chem. SAR descriptors employing sequential multiple regression analysis method. The models also provide valuable insight into the mechanism of action of these compounds. The developed models showed good statistical significance in internal (r2, q2 group cross validation and bootstrapping) validation and performed very well in predicting the Biological Activity (BA) of the compounds in the test set.
ACKNOWLEDGMENT
The authors are thankful to Head, School of Pharmacy, Devi Ahilya Vishwavidyalaya Indore India to provide trial version of software.
REFERENCES
- De Souza, M.V., 2006. Promising drugs against tuberculosis. Recent Pat. Antiinfect. Drug Discov., 1: 33-44.
PubMed - De Souza, M.V., 2006. Current status and future prospects for new therapies for pulmonary tuberculosis. Curr. Opin. Pulm. Med., 12: 167-171.
PubMed - Dye, C., B.G. Williams, M.A. Espinal and M.C. Raviglions, 2002. Erasing the worlds slow stain: Strategies to beat multidrug-resistant tuberculosis. Science, 295: 2042-2046.
CrossRefDirect Link - Dye, C. and B.G. Williams, 2000. Criteria for the control of drugresistant tuberculosis. Proc. Natl. Acad. Sci. USA., 97: 8180-8185.
PubMed - Desai, B., D. Sureja, Y. Naliapara, A. Shah and A.K. Saxena, 2001. Synthesis and QSAR Studies of 4-Substituted phenyl-2,6-dimethyl-3, 5-bis-N-(substituted phenyl)carbamoyl-1,4-dihydropyridines as potential antitubercular agents. Bioorg. Med. Chem., 9: 1993-1998.
Direct Link - El-Sayed, K.A., P. Bartyzel, X.Y. Shen, T.L. Perry, J.K. Zjawiony and M.T. Hamann, 2000. Marine natural products as antituberculosis agents. Tetrahedron, 56: 949-953.
CrossRef - Fujita, T. and T. Ban, 1971. Structure-activity relation. 3. Structure-activity study of phenethylamines as substrates of biosynthetic enzymes of sympathetic transmitters. J. Med. Chem., 14: 148-152.
CrossRef - Gupta, A.K., B.M. Arockia and S.G. Kaskhedikar, 2004. VALSTAT : Validation program for quantitative structure activity relationship studies. Indian J. Pharm. Sci., 66: 396-402.
Direct Link - Gupta, S.P. and S. Kumaran, 2006. Quantitative structure-activity relationship studies on benzodiazepine hydroxamic acid inhibitors of matrix metalloproteinases and tumor necrosis factor-α converting enzyme. Asian J. Biochem., 1: 47-56.
CrossRefDirect Link - Chaudhary, G., C. Karthikeyan, N.S. Hari Narayana Moorthy and T. Piyush, 2008. Quantitative structure activity relationship analysis of some diarylsulphonylurea derivatives as tubulin binding agents. Int. J. Cancer Res., 4: 1-11.
CrossRefDirect Link - Gokhale, V.M. and V.M. Kulkarni, 2000. Understanding the antifungal activity of terbinafine analogues using quantitative structure-activity relationship (QSAR) models. Bioorg. Med. Chem., 8: 2487-2499.
PubMed - Ghosh, P. and M.C. Bagchi, 2010. Anti-tubercular drug designing by structure based screening of combinatorial libraries. J. Mol. Model.
CrossRefDirect Link - Manvar, A.T., R.R.S. Pissurlenkar, V.R. Virsodia, K.D. Upadhyay and D.R. Manvar et al., 2010. Synthesis, in vitro antitubercular activity and 3D-QSAR study of 1,4-dihydropyridines. Mol. Divers., 14: 285-305.
CrossRef - Heym, B. and S.T. Cole, 1997. Multidrug resistance in Mycobacterium tuberculosis. Int. J. Antimicrob. Agents, 8: 61-70.
Direct Link - Karthikeyan, C., D. Santosh, N.S.H.N. Moorthy and P. Trivedi, 2007. QSAR analysis of HIV-1 reverse transcriptase inhibitory 5-Alkyl-2-[(Aryl and Alkyloxylcarbonylmethyl) Thio]-6-(1-Napthylmethyl) Pyrimidin-4 (3H)-Ones. Int. J. Virol., 3: 19-27.
CrossRefDirect Link - Karthikeyan, C., P.M. Kumar, N.S.H.N. Moorthy, S.K. Shrivastava and T. Piyush, 2006. Quantitative structure activity relationships of some selective inhibitors of glucagon receptor: A hansch approach. Asian J. Biochem., 1: 307-315.
CrossRefDirect Link - Mahapatra, A., S.P.N. Mativandlela, B. Binneman, P.B. Fourie and C.J. Hamilton et al., 2007. Activity of 7-methyljuglone derivatives against Mycobacterium tuberculosis and as subversive substrates for mycothiol disulfide reductase. Bioorg. Med. Chem., 15: 7638-7646.
CrossRef - Moorthy, N.S.H.N. and P. Trivedi, 2006. QSAR modeling of some 2-methoxy Acridones: Cytotoxic agents in multi drug resistant cells. Int. J. Cancer Res., 2: 267-276.
CrossRefDirect Link - Narute, A.S., P.B. Khedekar and K.P. Bhusari, 2008. QSAR studies on 4-thiazolidinones and 2-azetidinones bearing benzothiophene nucleus as potential anti-tubercular agents. Indian J. Chem., 47B: 586-591.
Direct Link - Pablos-Mendez, A., D.K. Gowda and T.R. Frieden, 2002. Controlling multidrug-resistant tuberculosis and access to expensive drugs: A rational framework. Bull. World Health Organ., 80: 489-495.
PubMed - Petrini, B. and S. Hoffner, 1999. Drug-resistant and multidrug-resistant tubercle bacilli. Int. J. Antimicrob. Agents, 13: 93-97.
Direct Link - Sharma, S., M.C. Sharma and A.D. Sharma, 2010. Quantitative structure-activity relationship analysis of some 2- substituted halogenbenzimidazoles analogues using computer-aided drug designing technique. J. Chem. Pharm. Res., 2: 357-365.
Direct Link - Sivakumar, P.M., S.P. Seenivasan, V. Kumar and M. Doble, 2007. Synthesis, antimycobacterial activity evaluation and QSAR studies of chalcone derivatives. Bioorg. Med. Chem. Lett., 17: 1695-1700.
CrossRef - Tomioka, H., 2002. Prospects for development of new antituberculous drugs. Kekkaku, 77: 573-584.
PubMed - Telenti, A. and M. Iseman, 2000. Drug-resistant tuberculosis: What do we do now. Drugs, 59: 171-179.
PubMed