
Zeinab A. Abd Elhaleem
Forensic Medicine and Clinical Toxicology, Faculty of Medicine, Ain Shams University, Egypt
Ahmed Elsayed
Internal Medicine, National Research Centre, Egypt
Journal of Pharmacology and Toxicology
Year: 2011 | Volume: 6 | Issue: 3 | Page No.: 258-271







ABSTRACT
Statins are the mainstay in the pharmacological management of dyslipidemia. They may also have anti-inflammatory, anti-proliferative and anti-oxidative effects. Since they are widely prescribed, their safety remains an issue of concern. The major untoward effect of statins is myotoxicity, which ranges from mild to severe. In addition to inhibiting endogenous cholesterol synthesis, however, statins decrease coenzyme Q10 (CoQ10) synthesis, the decrease of which may be implicated in statin myotoxicity. Hence, the present study evaluated the effect of CoQ10 supplementation against simvastatin and atorvastatin induced myotoxicity, measuring creatinine phosphokinase (CPK), lactate dehydrogenase (LDH), myoglobin as well as K, creatinine and CoQ10 level along with histopathological determinants. Sprague-dawley adult male albino rats were orally administered simvastatin or atorvastatin (20, 80 mg day-1) for 3 months with concurrent oral CoQ10 (100 mg day-1) administration. Simvastatin or atorvastatin in the higher dose used caused significant elevation of CPK, LDH, K and myoglobin, depletion of CoQ10 level along with histological muscle damage. This was more evident with atorvastatin with respect to simvastatin. Concurrent administration of CoQ10 showed significant modulation of CPK, LDH, K and myoglobin concomitant to restoration of depleted CoQ10 level and improvement in histopathological muscle findings. Present findings demonstrate the protective effect of CoQ10 against myotoxicity of simvastatins and atorvastatin suggesting its use to avoid poor adherence to statin treatment and increase clinical benefit.
PDF Abstract XML References Citation
Received: October 09, 2010;
Accepted: January 12, 2011;
Published: February 07, 2011
How to cite this article
Zeinab A. Abd Elhaleem and Ahmed Elsayed, 2011. Coenzyme Q10 Ameliorates Statin-related Myotoxicity: A Biochemical and Histological Study. Journal of Pharmacology and Toxicology, 6: 258-271.
DOI: 10.3923/jpt.2011.258.271
URL: https://scialert.net/abstract/?doi=jpt.2011.258.271
DOI: 10.3923/jpt.2011.258.271
URL: https://scialert.net/abstract/?doi=jpt.2011.258.271
INTRODUCTION
3-Hydroxy- 3-methyl glutaryl-coenzyme A (HMG-CoA) reductase inhibitors (‘statins’) represent the most effective and widely prescribed drugs currently available for the reduction of low-density lipoprotein cholesterol, a critical therapeutic target for primary and secondary prevention of cardiovascular atherosclerotic disease (Jeevan Shetty et al., 2008; Yiannis et al., 2010).
Although statins are safe and well-tolerated drugs, one of their most important clinical untoward effects is myotoxicity, ranging from mild myopathy to serious rhabdomyolysis (Itagaki et al., 2009). This risk has been emphasized by the withdrawal of cerivastatin in August 2001 after the drug was associated with approximately 100 rhabdomyolysis-related deaths (Fuhrmans, 2002). Rhabdomyolysis was also a factor in the decision by Merck and Co to abandon the development of a 160 mg sustained-release simvastatin formulation in the mid-1990s (Davidson et al., 1997).
Based on recent evidence in the cardiovascular literature, many authors are advocating more aggressive therapy for hyperlipidemia (Murphy et al., 2009; Nicholls et al., 2010) a strategy that will likely lead to higher doses of statin therapy and the use of the most potent and possibly muscle-unfriendly statins (Radcliffe and Campbell, 2008).
Recently, the pleiotropic effects of statins as antioxidant, anti-inflammatory and antiproliferative have emerged (Anand et al., 2008; Hajipour et al., 2010). This led to expansion of their use in other disorders, such as cancer, stroke, inflammatory conditions and polycystic ovarian syndrome (Kishi et al., 2009; Sathyapalan and Atkin, 2010; Vasiuk et al., 2010). This place more patients at risk of myotoxicity. Clinically, mild myopathic symptoms may lead to poor adherence to statin treatment whereas extreme myopathy manifests as the rare but life-threatening event rhabdomyolysis (Peters et al., 2009).
Moreover, it is quite easy to miss clinically significant weakness unless the clinician is suspicious and uses proper strength-assessment techniques. Only patients with weakness severe enough to interfere with activities of daily living are likely to be recognized as suffering from statin-induced muscle weakness (Radcliffe and Campbell, 2008).
Little is known regarding how statins produce muscle injury, but several theories have been proposed based on the biosynthetic pathways inhibited by statins. One of these theories is that statin therapy inhibits production of mevalonate which is a precursor of many substances necessary for maintenance of the cell wall and is also an intermediary metabolite in the synthesis of coenzyme Q10 (ubiquinone) the inhibition of which may explain some of the myotoxic effects of statins (Liu et al., 2010).
Coenzyme Q10 (CoQ10) a 1, 4-benzoquinone with a 50-carbon isoprenoid side chain, was first isolated from beef heart mitochondria by Frederick Crane of Wisconsin, USA, in 1957. It is fat soluble and approximately 50% of its body's content is thought to be obtained from the diet, with meat products being the largest source in the normal diet, whereas 50% is derived from endogenous synthesis (Weber et al., 1997).
CoQ10 is an essential component of the mitochondrial electron transport system and its deficiency may affect oxidative phosphorylation and mitochondrial adenosine triphosphate (ATP) production. Therefore, CoQ10 deficiency resulting from statin treatment may impair muscle energy metabolism and contribute to the development of myopathy and muscle symptoms, described in patients treated with statins (Caso et al., 2007). Hence, this study was undertaken to assess for the possible myotoxic effect of the two commonly prescribed statins, simvastatin and atorvasatatin in Sprague-dawley adult male albino rats and whether supplementation with CoQ10 would improre this.
MATERIALS AND METHODS
Animals: Seventy seven Sprague-dawley adult male albino rats weighting 150-200 g were maintained under controlled temperature (22-25°C) and 12 h alternate light and dark cycle. They were locally bred at the animal house in the Medical Research Center, Faculty of Medicine, Ain Shams University in 2009. Standard laboratory animal feed and water were given ad libitum. Rats were acclimatized to experimental conditions prior to the start dosing for a period of one week. All experiments were conducted in accordance with the guidelines for the use of laboratory animals.
Drugs, dosage and route of administration Simvastatin, one of the first generation statin supplied from Merck and Co. Company of pharmacy, Egypt in the form of tablets of 20 mg.
Atorvastatin, one of the second generation statin supplied from Egyptian International Pharmaceutical Industries Company (EPICO), company of pharmacy, Egypt in the form of tablets of 20 mg.
Each drug was dissolved in distilled water and given as suspension by gavage. Each rat received simvastatin or atorvastatin in a dose of 0.36 or 1.44 mg day-1. These doses are equivalent to adult human therapeutic dose of 20 and 100 mg day-1, respectively.
The lower dose of simvastatin and atorvastatin was chosen on the basis of data present in the literature (Jula et al., 2002; Phillips et al., 2002).
For both drugs the higher dose used was the Maximum Tolerated Dose (MTD). It was selected according to Murphy et al. (2009) and Nicholls et al. (2010) who recommend this dose for greater lipid goal achievement and more effective cardiovascular prevention.
Coenzyme Q10 was supplied from Arab Company for Pharmaceuticals and Medicinal Plants, MEPACO-Egypt in the form of capsules of 30 mg. It was dissolved in distilled water and given as suspension by gavage. Each rat received 1.8 mg day-1. This dose is equivalent to adult human dose of 100 mg day-1. It was selected according to Caso et al. (2007).
Animal groups: The animals were divided into eleven experimental groups (7 rats each) as follows: the first group was untreated control group, the second only with the vehicle (distilled water) used to dissolve the drugs, the third was treated with 100 mg day-1 of CoQ10, the fourth with low dose simvastatin (20 mg day-1), the fifth with MTD simvastatin (80 mg day-1), the sixth with low dose atorvastatin (20 mg day-1), the seventh with MTD atorvastatin (80 mg day-1), the eights with CoQ10 and low dose simvastatin, the ninth with CoQ10 and MTD simvastatin, the tenth with CoQ10 and low dose atorvastatin, the eleventh with CoQ10 and MTD atorvastatin. The test groups and their control were kept on prescribed treatment for three months.
The results obtained from the three control groups were similar, so we have combined them and show the results as a single control value.
During the treatment, changes in body weight of rats have been recorded weekly. The performance of skeletal muscle system was evaluated by testing the righting reflex daily in each rat (the ability of the rat to straighten itself on four legs when turned on its back), alteration of which represents a sign of myotonic disorder.
Biochemical analysis At the end of the experimental period, 24 h urinary samples were collected for determination of urinary myoglobin and creatinine by using standard techniques. Then the rats were anesthetized with thiopental Na, the abdomen was dissected and blood samples from the aorta were collected in ethylenediaminetetracetic acid (EDTA) rinsed tubes and then centrifuged at 600 g for 10 min. The plasma was separated and stored at -20°C until assay. Creatinine phosphokinase (CPK), lactic acid dehydrogenase (LDH), potassium (K), myoglobin and creatinine determination were assayed by using standard techniques.
Measurement of plasma CoQ10: The collected plasma samples were stored in a -75°C freezer. Then, they were thawed one at a time for CoQ10 analysis according to the method of Edlund (1988). In brief, 1 mL of n-propanol was added to 300 μL of plasma or standard CoQ10 solution. After vigorous mixing the mixture was left to stand for 3 min, followed by remixing and centrifugation at 2500 g for 5 min. A supernatant volume of 80 μL was immediately injected for High Performance Liquid Chromatographic analysis (HPLC) of CoQ10.
Histological examination:
Light microscopic examination: After collection of blood samples, gastrocnemius muscles were dissected from origin to insertion in all rat groups. Every muscle was dissected out and fixed in 10% formal saline. Paraffin sections about 5u thick were prepared from each block and stained with hematoxylin and eosin (H and E) (Drury and Wallington, 1983).
Electron microscopic examination: Small parts of muscles (1 mm2), were fixed in formal gluteraldehyde to be processed for electron microscopic study. After being embedded, ultra-thin sections (60-90 nm) were cut using the ultramicrotome, mounted on copper grids and stained with uranyl acetate and lead citrate (Reynolds, 1963). Stained grids were examined using transmission electron microscopy at 60-80 kilo electron volt.
Statistical analysis: The obtained results were expressed as mean±standard deviation followed by student t-test analysis where p<0.05 was considered statistically significant.
RESULTS
Effect of chronic treatment with simvastatin, atorvastatin, CoQ10+simvastatin or atorvastatin on animal health: Rats chronically treated with MTD simvastatin or atorvastatin (80 mg day-1) showed adverse physical signs such as a decrease in food consumption and a significant decrease in body weight (Fig. 1). One out of 7 rats treated with MTD atorvastatin showed slowing of the righting reflex. This rat died before the end of the treatment. The remaining rats were examined for biochemical and histological parameters at the end of the treatment.
In contrast, rats treated with the lower doses of simvastatin and atorvastatin as well as those given CoQ10 and simvastatin or atorvastatin showed normal body weight and appeared in good health similarly to control rats. In all these animals, the righting reflex was normal during the whole treatment period.
Effect of chronic treatment with simvastatin, atorvastatin, CoQ10+simvastatin or atorvastatin on biochemical parameters of rat plasma: Plasma level of CPK, LDH and myoglobin as well as K and creatinine level were measured. Non significant difference was observed in all tested parameters in low dose simvastation, atorvastatin, CoQ10+simvastatin or atorvastatin treated groups compared to control group, p>0.05.
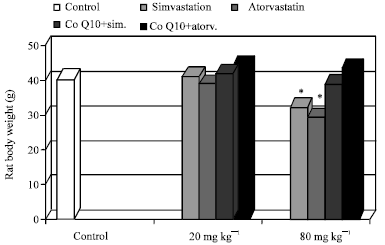 | |
| Fig. 1: | Effect of chronic treatment with simvastarin, atorvastatin and Co10+simvastarin or atorvastatin on rat body weight. Values are presented as Mean±SD, *p<0.05 when compared to control |
In plasma of MTD (80 mg day-1) simvastatin and atorvastatin treated rats we found non significant difference in creatinine level compared to control group. A significant increase in CPK, LDH, myoglobin and K as signs of marked muscle involvement was observed. This increase was more obvious with atorvastatin group, p<0.001 than simvastatin group, p<0.05. Administration of CoQ10 induced restoration of all tested parameters to normal control value in MTD simvastatin treated group, p>0.05 while it is still significant with control in MTD atorvastatin treated group, p<0.05 (Table 1).
Effect of chronic treatment with simvastatin, atorvastatin, CoQ10+ simvastatin or atorvastatin on urinary myoglobin and creatinine: Simvastatin and atorvastatin treated rats with MTD (80 mg day-1) showed significant increase of myoglobinuria. Concurrent administration of CoQ10 with MTD simvastatin and atorvastatin decreased the elevated level of urinary myoglobin to control value in MTD simvastatin treated rats only, p>0.05. In all other treated groups, this parameter was unchanged. Also, no modification of creatinine level in urine was found in all tested groups (Table 2).
| Table 1: | Effect of chronic treatment with simvastatin, atorvastatin, CoQ10+simvastatin or atorvastatin on plasma CPK, LDH, K, myoglobin and creatinine |
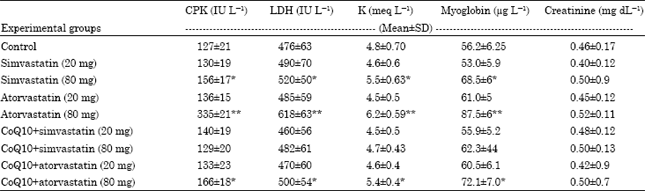 | |
| CPK, creatinine phosphokinase; LDH, lactic acid dehydrogenase: K, potassium; Values are Mean±SD, *p<0.05 when compared to control, **p<0.001 when compared to control | |
| Table 2: | Effect of chronic treatment with simvastatin, atorvastatin, CoQ10+ simvastatin or atorvastatin on urinary myoglobin and creatinine |
 | |
| Values are Mean±SD, *p<0.05 when compared to control, **p<0.001 when compared to control | |
Effect of chronic treatment with simvastatin, atorvastatin, CoQ10+simvastatin or atorvastatin on plasma CoQ10 level: Low dose and MTD simvastatin and atorvastatin resulted in significant decrease in plasma CoQ10 compared to control group. This decrease was more significant with MTD simvastatin and atorvastatin, p<0.001 than with lower doses of both drugs, p<0.05.
Administration of CoQ10 with lower doses of simvastatin or atorvastatin and MTD of simvastatin restored the depleted level of plasma CoQ10 to normal control value, p>0.05. In contrast its administration with MTD of atorvastatin increased depleted plasma CoQ10 but not to control value, p<0.05 (Fig. 2).
Effect of chronic treatment with simvastatin, atorvastatin, CoQ10+simvastatin or atorvastatin on histology of rat skeletal muscle: The observations were graded as (-) nil, (+) mild, (++) moderate and (+++) severe on the basis of severity and appearance of histological changes such as degeneration, inflammatory cells and vacuolation of sarcoplasm and mitochondria (Table 3). The skeletal muscle of the control group showed normal histology (Fig. 3 and 4). Light microscopic examination of skeletal muscle fibers of low dose of both drugs showed nearly normal control appearance.
 | |
| Fig. 2: | Effect of chronic treatment with simvastarin, atorvastatin and Co10+simvastarin or atorvastatin on plasma Q10 level.Values are Mean±SD, *p<0.05 when comared to control, **p<0.01 when comared to control |
| Table 3: | Histopathological changes of rat skeletal muscles in control, simvastatin, atorvastatin and CoQ10+simvastatin or atorvastatin groups |
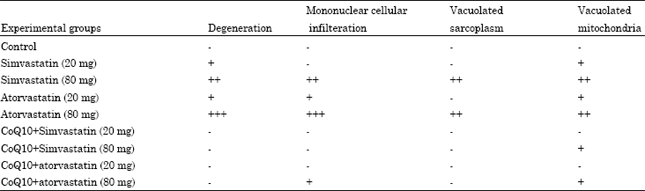 | |
| - = Nil, + = Mild, ++ = Moderate, +++ = Severe | |
 | |
| Fig. 3: | A photomicrograph of rat skeletal muscle showing normal muscle cells (control group). H&E x 640 |
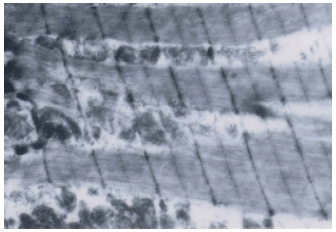 | |
| Fig. 4: | An electron micrograph of a portion of rat skeletal myofibril of control group. x 6000 |
On the other hand electron microscopic examination of low dose simvastatin treated rats, showed few areas of myofibrillar loss and few mitochondria with vacuolation (Fig. 5). Low dose atorvastatin treated group showed degenerative changes, myofibrillar loss and mitochondrial vacuolation (Fig. 6). Administration of MTD simvastatin resulted in vacuolation of sarcoplasm of some skeletal muscle cells, degeneration with mononuclear cellular infiltration (Fig. 7). These findings were more pronounced with MTD atorvastatin as there was a more profound infiltration by mononuclear cells, with wide separation of muscle fibers and vacuolated sarcoplasm (Fig. 8).
CoQ10 adminstration, ameliorated simvastatin and atorvastatin myotoxicity manifested by improvement in previous histological findings to normal or near normal (Fig. 9 and 10). In CoQ10 and MTD atorvastatin treated group, electron microscopic examination, revealed normal myofibrils apprearance with minimal mitochondrial vacuolation.
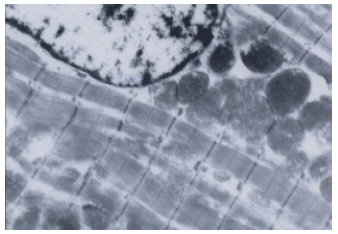 | |
| Fig. 5: | Electron microscopic examination of skeletal muscle fibers of low dose simvastatin group, showing few areas of myofibrillar loss and few mitochondria with vacuolation . x 6000 |
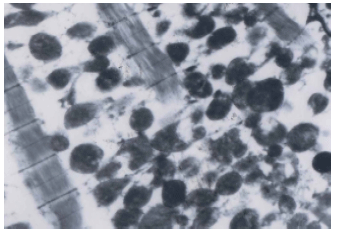 | |
| Fig. 6: | Electron microscopic examination of skeletal muscle fibers of low dose atorvastatin group, showing degenerative changes with myofibrillar loss and occasional mitochondrial vacuolation. x 10000 |
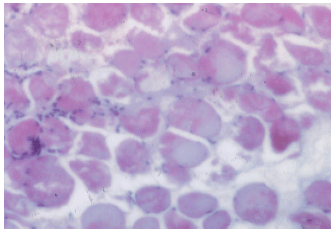 | |
| Fig. 7: | A photomicrograph of skeletal muscle fibers of MTD simvastatin group, showing, degeneration of some muscle cells, others show vacuolated sarcoplasm with mononuclear cellular infiltration. H&E x 250 |
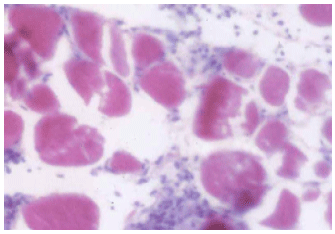 | |
| Fig. 8: | A photomicrograph of skeletal muscle fibers of MTD atorvastatin group, showing heavy infiltration by mononuclear cells, with wide separation of muscle fibers and vacuolated sarcoplasm. H&E x 250 |
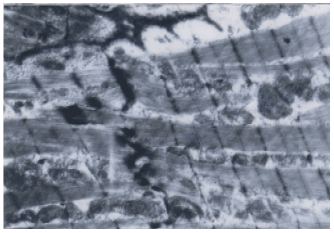 | |
| Fig. 9: | Electron microscopic examination of skeletal muscle fibers of CoQ10+MTD simvastatin group, showing nearly normal myofibrils appearance |
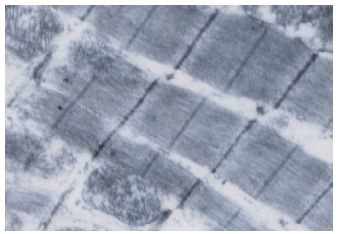 | |
| Fig. 10: | Electron microscopic examination of skeletal muscle fibers of CoQ10+MTD atorvastatin group,showing normal myofibrils appearance with minimal mitochondrial vacuolation. x 15000 |
DISCUSSION
Treatment with statin drugs has been associated with a variety of Skeletal muscle-related complaints ranging from myalgia and cramps to exercise intolerance, weakness and occasionally acute muscle breakdown with rhabdomyolysis (Lamperti et al., 2005).
In the present study, despite depletion of CoQ10 level and presence of histopathological muscle changes with low dose simvastatin and atorvastatin (20 mg day-1), the plasma level of CPK, LDH, K, myoglobin, creatinine as well as urinary myoglobin and creatinine showed non significant difference compared to control. The presence of normal CPK level despite histopathological skeletal muscle findings coincides with the study of Phillips et al. (2002).The authors reported normal CPK level in patients on statin therapy with muscle pain, weakness and histopathological findings of myopathy. They proposed that this may represent muscle toxicity below the threshold needed to increase parameters of muscle injury. Mohaupt et al. (2009) added that this may be due to restriction of muscle fiber damage to the intracellular space. Hence, the intact sarcolemma likely prevents leakage of CPK into the bloodstream.
In contrast, MTD simvastatin and atorvastatin increased the plasma level of CPK, LDH, K, myoglobin and urinary myoglobin, although not much as during rhabdomyolysis. These may represent early markers of myopathy occurring before rhabomyolysis as muscle injury leads to release of CPK, LDH and myoglobin into blood stream with subsequent urinary excretion of myoglobin (Pierno et al., 2006).
On the other hand, non significant difference in creatinine level was observed. The presence of normal creatinine level despite significant myoglobinuria could be attributed to the degree of muscle damage as greater degree of muscle damage and CPK elevations are usually required to cause myoglobin-pigment induced renal dysfunction. Moreover, Thompson et al. (2003) stated that renal problems may not be related to the degree of muscle injury alone but other factors may contribute such as, the patient's hydration status, concomitant medication or genetic predisposition.
This was confirmed by histological findings in the form of degenerative changes with myofibrillar loss, sarcoplasmic and mitochondrial vacuolation, with mononuclear cellular infiltration. These findings were more pronounced with MTD atorvastatin. Findings of the present study are in agreement with those of Radcliffe and Campbell (2008) who reported that the muscle pathology in statin myopathy is most often nonspecific, with fiber necrosis, degeneration, regeneration and phagocytic infiltration. The development of vacuoles in the current study has also been reported as characteristic morphological abnormality in statin-treated animals and human skeletal muscles (Chucrallah et al., 1992; Bergman et al., 2003). This suggests that the statins may have disturbed intracellular membrane vesicle transport in the myofibers.
The presence of intensified muscle damage with MTD atorvastatin could be attributed to the difference in metabolic properties of the individual drug used. Atorvastatin is given in the active acid form. In contrast, simvastatin is administered in inactive form as lactones, some portion of which is then hydrolyzed into the active acid form to produce their clinical effects. In terms of elimination from the body, atorvastatin has an approximate elimination half-life of 14 h. Noteworthy, its HMG-CoA reductase inhibitory activity appears to have a half-life of 20-30 h, which is thought to be due to its active metabolites. On the other hand half life of simvastatin is about 2-3 h. This suggests that drug accumulation may also contribute to increased risk of myotoxicity (Bellosta et al., 2004).
Our results agree with those of Pierno et al. (2006) who found that atorvastatin was more potent than simvastatin in modifying muscle function. They found that administration of atorvastatin at doses lower than simvastatin induced a marked reduction of the muscle resting chloride conductase with a concomitant increase of sarclemma excitability and shifted mechanical threshold for contraction towards more negative potentials.
A variety of hypothesis has been suggested to account for the myotoxic effects of statins. Blocking cholesterol synthesis is one of these hypotheses. Because membrane lipids are in dynamic equilibrium with plasma lipids, a low concentration of plasma cholesterol associated with decreased intracellular cholesterol may result in reduced membrane lipid content, which in turn may cause physical modification of membrane fuidity and a decrease in cell proliferation (Morita et al., 1997). Such changes in membrane composition may result in impaired membrane Na-K channel function, thus causing irreversible cell damage (Lijnen et al., 1994). However, blocking cholesterol synthesis with squalene synthase inhibitors, does not produce myotoxicity in vitro models, suggesting that other compounds produced by HMG-CoA reductase activity are responsible (Flint et al., 1997).
Statins and HMG-CoA reductase inhibition block production of farnesyl pyrophosphate and geranyl geranyl pyrophosphate, intermediary for the production of CoQ10 (Thompson et al., 2003). CoQ10 is an essential cofactor in mitochondrial oxidative phosphorylation and is necessary for ATP production. In this role, CoQ10 acts as a mobile electron carrier, transferring electrons from complex I (NADH CoQ reductase) to complex III (cytochrome bc1 complex) or from complex II (succinate dehydrogenase) to complex III. Depletion of CoQ10 leads to mitochondrial impairment with increased mitochondrial Ca permeability and Ca release from sarcoplasmic reticulum (Molyneux et al., 2008). Both mitochondrial and calcium impairments may account for apoptosis process, oxidative stress and muscle remodeling and degeneration that have been extensively reported to explain statin myotoxicity and functional symptoms described by statin-treated patients (Sirvent et al., 2008).
There is other evidence that supports a role for CoQ10 depletion in producing statin myopathy. The ratio of lactate to pyruvate is higher in statin-treated patients, suggesting a shift toward anaerobic metabolism and possible mitochondrial dysfunction (Thompson et al., 2003).
In the current study, plasma level of CoQ10 was significantly depleted following lower and MTD of simvastatin and atorvastatin. Concurrent administration of CoQ10 resulted in restoration of plasma Q10 level to normal control value in low dose of both drugs and MTD of simvastatin only. Moreover, concomitant to decreased CPK, LDH, K and myoglobin levels, an improvement in histopathological muscle findings was observed.
Depletion of plasma CoQ10 level in the present study coincides with previous studies that have demonstrated reduction of plasma CoQ10 levels in the course of statin treatment (Marcoff and Thompson, 2007; Villalba et al., 2010). This depletion was dose dependant and improved with CoQ10 supplementation. This agrees with Molyneux et al. (2008) who stated that the magnitude of statin induced depletion of CoQ10 levels may be drug and dose related and that supplementation with oral CoQ10 can restore plasma CoQ10 levels in patients receiving statin therapy.
Furthermore, 40% reduction in myopathic pain and 38% reduction in pain interference with daily activities after 30 days of 100 mg day-1 CoQ10 supplementation compared with no change following 400 mg IU day-1 of vitamin E in patients with statin related myopathy on concurrent statin treatment was reported by Caso et al. (2007).
The protective effect of CoQ10 may be due to compensation of depleted Q 10 level or its antioxidant effect (Molyneux et al., 2008). Moreover, oxidized CoQ10 is reduced to CoQ10H2 in the mitochondria by flavoenzymes including mitochondrial succinate dehydrogenase and NADH 3 dehydrogenase. The reduced form of CoQ10 is also antioxidant and is the only endogenously synthesized lipophilic antioxidant. Hence, it can act as antioxidant directly, protecting biological membranes against oxidation (Littarru and Tiano, 2010; Villalba et al., 2010). As CoQ10 is lipophilic and transported in lipoprotein particles in the circulation, it can inhibit the peroxidation of lipoprotein lipids present in the circulation. It can also have a role in recycling α tocopherol as reviewed by Sohal (2004).
In addition, CoQ10 affects expression of genes involved in human cell signaling, metabolism, and transport and some of the effects of exogenously administered CoQ10 may be due to this property (Littarru and Tiano, 2007).
Finally, recent data pointed that CoQ10 could have a direct effect on endothelial function. Supplementation with CoQ10 significantly affects endothelium-bound- extracellular SOD activity with improvement in flow-dependent endothelial-mediated dilatation, a functional parameter commonly used as a biomarker of vascular function (Littarru and Tiano, 2010).
In conclusion, the results of the present study showed that the myotoxic effect of statin was drug and dose dependant. Myotoxicity was more evident with atorvastatin with respect to simvastatin. Our findings suggest that a normal CPK level does not rule out muscle pathology. Prevention is the best approach to managing statin-related myopathy. This includes using the lowest statin dose required to achieve therapeutic goals. Also, Patients must understand the risk of statin therapy and be willing to promptly report any untoward reactions. Supplementation with CoQ10 ameliorated statin-related myotoxic effect suggesting its use to avoid poor adherence to statin treatment and increase clinical benefit. Whether CoQ10 supplementation might improve statin induced myotoxicity by increased depleted Q10 level or oxidative phosphorylation in mitochondria merits further investigation.
ACKNOWLEDGMENT
We express our sincere thanks to Dr. Seham Kamel, Department of histology, Faculty of Medicine, Ain Shams University for her valuable help in the histological part of the study. We are also grateful to Dr. Emad Ibrahim, Department of Biochemistry, Faculty of Science, Ain Shams University for his assistance with the plasma Q10 level assay.
REFERENCES
- Anand, V.A., M. Chenniappan, S. Kalavathy, K. Uma, M.P. Saravanan and S. Kumar, 2008. Redeeming measure of atorvastatin in the risk factors of cardiovascular disease. Int. J. Pharmacol., 4: 305-309.
CrossRefDirect Link - Bellosta, S., R. Paoletti and A. Corsini, 2004. Safety of statins: Focus on clinical pharmacokinetics and drug interactions. Circulation, 109: III-50-III-57.
CrossRefPubMedDirect Link - Bergman, M., H. Salman, M. Djaldetti, S. Alexandrova, I. Punsky and H. Bessler, 2003. Ultrastructure of mouse striated muscle fibers following pravastatin administration. J. Muscle Res. Cell Motil., 24: 417-420.
CrossRef - Caso, G., P. Kelly, M.A. McNurlan and W.E. Lawson, 2007. Effect of coenzyme Q10 on myopathic symptoms in patients treated with statins. Am. J. Cardiol., 99: 1409-1412.
CrossRefPubMedDirect Link - Chucrallah, A., U. De Girolami, R. Freeman and M. Federman, 1992. Lovastatin/Gemfibrozil myopathy: A clinical, histochemical and ultrastructural study. Eur. Neurol., 32: 293-296.
PubMed - Davidson, M.H., E.A. Stein, C.A. Dujovne, B. Amin and M. Tobert, 1997. The efficacy and six-week tolerability of simvastatin 80 and 160 mg/day. Am. J. Cardiol., 79: 38-42.
CrossRef - Edlund, P.O., 1988. Determination of coenzyme Q-10, a-tocopherol and cholesterol in biological samples by coupled column LC with coulometric an ultraviolet detection. J. Chromatogr. B Biomed. Sci. Appl., 425: 87-97.
CrossRef - Hajipour, B., M.H. Somi, F. Dibazar, N.A. Asi and A.M. Vatankhah, 2010. Anti-oxidative effect of simvastatin in liver and lung tissue after hepatic ischemia/reperfusion in rat. J. Med. Sci., 10: 66-70.
CrossRefDirect Link - Itagaki, M., A. Takaguri, S. Kano, S. Kaneta, K. Ichihara and K. Satoh, 2009. Possible mechanisms underlying statin-induced skeletal muscle toxicity in L6 fibroblasts and in rats. J. Pharmacol. Sci., 109: 94-101.
PubMed - Shetty, J.K., M. Prakash, S. Tripathy, M. Verma and P. Vikram, 2008. Effect of atorvastatin on paraoxonase activity in-patients with hyperlipidemia. Asian J. Biochem., 3: 139-142.
CrossRefDirect Link - Kishi, T., A. Yamada, S. Okamatsu and K. Sunagawa, 2009. Atorvastatin might improve ventricular electrostability and decelerate the deterioration of renal function in patients with heart failure and diabetes mellitus. J. Cardiol., 53: 341-348.
CrossRef - Lamperti, C., A.B. Naini, V. Lucchini, A. Prelle and N. Bresolin et al., 2005. Muscle coenzyme Q10 level in statin-related myopathy. Arch. Neurol., 62: 1709-1712.
PubMed - Lijnen, P., H. Celis, R. Fagard, J. Staessen and A. Amery, 1994. Influence of cholesterol lowering on plasma membrane lipids and cationic transport systems. J. Hypertens., 12: 59-64.
PubMed - Littarru, G.P. and L. Tiano, 2007. Bioenergetic and antioxidant properties of CoQ10: Recent developments. Mol. Biotechnol., 37: 31-37.
CrossRefDirect Link - Littarru, G.P. and L. Tiano, 2010. Clinical aspects of coenzyme Q10: An update. Nutrition, 26: 250-254.
CrossRefPubMedDirect Link - Liu, C.S., C.K. Lii, L.L. Chang, C.L. Kuo and W.L. Cheng et al., 2010. Atorvastatin increases blood ratios of vitamin E/low-density lipoprotein cholesterol and coenzyme Q10/low-density lipoprotein cholesterol in hypercholesterolemic patients. Nutr. Res., 30: 118-124.
CrossRef - Marcoff, L. and P.D. Thompson, 2007. The role of coenzyme Q10 in statin-associated myopathy: A systematic review. J. Am. Coll. Cardiol., 49: 2231-2237.
Direct Link - Mohaupt, M.G., R. Karas, E.B. Babiychuk, V. Sanchez-Freire and K. Monastyrskaya et al., 2009. Association between statin-associated myopathy and skeletal muscle damage. Can. Med. Assoc. J., 181: E11-E18.
CrossRefDirect Link - Molyneux, S.L., J.M. Young, C.M. Florkowski, M. Lever and P.M. George, 2008. Coenzyme Q10: Is there a clinical role and a case for measurement. Clin. Biochem. Rev., 29: 71-82.
Direct Link - Morita, I., I. Sato, L. Ma and S. Murota, 1997. Enhancement of membrane fluidity in cholesterol poor endothelial cells pre-treated with simvastatin. Endothelium, 5: 107-113.
PubMed - Murphy, S.A., C.P. Cannon, S.D. Wiviott, C.H. McCabe and E. Braunwald, 2009. Reduction in recurrent cardiovascular events with intensive lipid-lowering statin therapy compared with moderate lipid-lowering statin therapy after acute coronary syndromes from the PROVE IT-TIMI 22 (pravastatin or atorvastatin evaluation and infection therapy-thrombolysis in myocardial infarction 22) trial. J. Am. Coll. Cardiol., 54: 2358-2362.
CrossRef - Nicholls, S.J., G. Brandrup-Wognsen, M. Palmer and P.J. Barter, 2010. Meta-analysis of comparative efficacy of increasing dose of Atorvastatin versus Rosuvastatin versus Simvastatin on lowering levels of atherogenic lipids (from VOYAGER). Am. J. Cardiol., 105: 69-76.
CrossRef - Peters, B.J.M., O.H. Klungel, F.L. Visseren, A. de Boer and A.H.M. van der Zee, 2009. Pharmacogenomic insights into treatment and management of statin-induced myopathy. Genome Med., 1: 120-120.
PubMed - Phillips, P.S., R.H. Haas, S. Bannykh, S. Hathaway and R.N. Nancy et al., 2002. Statin associated myopathy with normal creatine kinase levels. Ann. Intern. Med., 137: 581-585.
Direct Link - Pierno, S., M.P. Didonna, V. Cippone, A. De Luca and M. Pisoni et al., 2006. Effects of chronic treatment with statins and fenofibrate on rat skeletal muscle: A biochemical, histological and electrophysiological study. Br. J. Pharmacol., 149: 909-919.
PubMed - Radcliffe, K. and W.W. Campbell, 2008. Statin myopathy. Curr. Neurol. Neurosci. Rep., 8: 66-72.
PubMed - Reynolds, E.S., 1963. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol., 17: 208-212.
CrossRefPubMedDirect Link - Sathyapalan, T. and S.L. Atkin, 2010. Evidence for statin therapy in polycystic ovary syndrome. Ther. Adv. Endocrinol. Metab., 1: 15-22.
CrossRef - Sirvent, P., J. Mercier and A. Lacampagne, 2008. New insights into mechanisms of statin-associated myotoxicity. Curr. Opin. Pharmacol., 8: 333-338.
PubMed - Thompson, P.D., P. Clarkson and R.H. Karas, 2003. Statin-associated myopathy. J. Am. Med. Assoc., 289: 1681-1690.
PubMedDirect Link - Vasiuk, I., I. Perlamutrov, M.N. Shkol`nik and E.L. Shkolnik, 2010. Possibilities of atorvastatin in complex management of extensive psoriasis in patients with arterial hypertension. Kardiologiia, 50: 37-46.
PubMed - Villalba, J.M., C. Parrado, M. Santos-Gonzalez and F.J. Alcain, 2010. Therapeutic use of coenzyme Q10 and coenzyme Q10-related compounds and formulations. Exp. Opin. Investig. Drugs, 19: 535-554.
CrossRefPubMedDirect Link - Weber, C., A. Bysted and G. Holmer, 1997. The coenzyme Q10 content of the average danish diet. Int. J. Vitam. Nutr. Res., 67: 123-129.
PubMedDirect Link - Yiannis, C.S., K.C. Konstantinos, M. Gesthimani, V. Christos, H.G. Apostolos and G.D. George, 2010. Risk factors and drug interactions predisposing to statin-induced myopathy: Implications for risk assessment, prevention and treatment. Drug Saf., 33: 171-187.
Direct Link