
Fazlul Huq
Discipline of Biomedical Science, School ofMedical Sciences,
Faculty of Medicine, C42, The University of Sydney, Lidcombe, NSW, Australia
Journal of Pharmacology and Toxicology
Year: 2008 | Volume: 3 | Issue: 2 | Page No.: 93-101







ABSTRACT
Zolpidem (ZP) is a new orally active sleep inducer belonging to the class of compounds known as imidazopyridine. Molecular modelling analyses based on molecular mechanics, semi-empirical (PM3) and DFT (at B3LYP/6-31G* level) calculations show that parent drug and all its metabolites have moderately large LUMO-HOMO energy differences so that none is expected to be highly labile kinetically. The molecular surface of ZP is found to abound in electron-deficient regions so that it can react with cellular nucleophiles such as glutathione and nucleobases in DNA, thus inducing cellular toxicity and causing DNA damage respectively. However, because of kinetic inertness, the rates of such adverse reaction may be low unless speeded up enzymatically. Increased incidence of nausea and vomiting associated with higher doses of ZP may be due to the parent drug rather than any of its metabolites.
PDF Abstract XML References Citation
How to cite this article
Fazlul Huq, 2008. Molecular Modelling Analysis of the Metabolism of Zolpidem. Journal of Pharmacology and Toxicology, 3: 93-101.
DOI: 10.3923/jpt.2008.93.101
URL: https://scialert.net/abstract/?doi=jpt.2008.93.101
DOI: 10.3923/jpt.2008.93.101
URL: https://scialert.net/abstract/?doi=jpt.2008.93.101
INTRODUCTION
Zolpidem (ZP; N,N,6-trimethyl-2-(4-methylphenyl)-imidazole[1,2-a]pyridine-3-acetamide L-(+)-hemitartrate) is a new orally active sleep inducer belonging the class of compounds known as imidazopyridine. It acts on the ω1-receptor subtype of the benzodiazepine binding site [Langtry and Benfield, 1990]. Since its introduction to clinic in the USA in the early nineties, ZP has become one of the most commonly used hypnotics in the elderly persons (Rush, 1998).
The drug is rapidly absorbed, has mean elimination half-life of about 2 h and is extensively metabolized and excreted as pharmacologically inactive metabolites. Only trace amounts of the unchanged drug is excreted. The biotransformation of ZP proceeds through different pathways including (I) methyl oxidation of the phenyl moiety to produce hydroxymethylphenylzolpiden (M1) and carboxyphenylzolpiden (M2), (ii) methyl oxidation of the imidazopyridine moiety to produce hydroxymethylimidazopyridinezolpiden (M3) and carboximidazopyridinezolpiden (M4) and (iii) hydroxylation of the imidazopyridine moiety to produce M5. Another minor pathway involves hydroxylation of one of the methyl groups of the substituted amide to produce M6. The metabolism of ZP is mediated by a number of cytochrome P450 isoenzymes including CYP3A4, CYP2A9, CYP1A2 and CYP2D6 (Von Moltke et al., 1999a,b). Because of its short elimination half-life, ZP has a low likelihood of producing residual sedative effects (Olubodun et al., 2003). The drug has little potential for abuse because higher doses are associated with increased incidence of nausea and vomiting (Debailleul et al., 1991).
In this study, molecular modelling analyses have been carried out using the program Spartan’ 04, 2004. to investigate the relative stability of ZP and its metabolites with the aim of providing a better understanding of their relative toxicity.
Computational methods
The geometries of ZP and its metabolites(Fig. 1) denoted as M1, M2, M3, M4, M5 and M6 have been optimised based on molecular mechanics, semi-empirical and DFT calculations, using the molecular modelling program Spartan ’04.
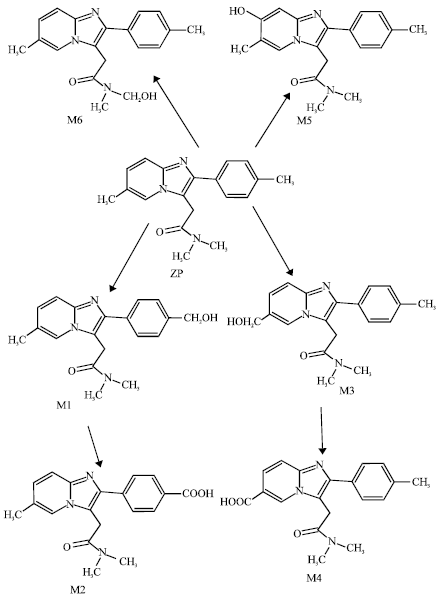 | |
| Fig. 1: | Metabolic pathways for ZP based on Hempel and Blaschke (1996) |
Molecular mechanics calculations were carried out using MMFF force field. Semi-empirical calculations were carried out using the routine PM3. DFT calculations were carried at B3LYP/6-31G* level. In optimization calculations, a RMS gradient of 0.001 was set as the terminating condition. For the optimised structures, single point calculations were carried out to give heat of formation, enthalpy, entropy, free energy, dipole moment, solvation energy, energies for HOMO and LUMO. The order of calculations: Molecular mechanics followed by semi-empirical followed by DFT ensured that the structure was not embedded in a local minimum. To further check whether the global minimum was reached, some calculations were carried out with improvable structures. It was found that when the stated order was followed, structure corresponding to the global minimum or close to that could ultimately be reached in all cases. Although RMS gradient of 0.001 may not be sufficiently low for vibrational analysis, it is believed to be sufficient for calculations associated with electronic energy levels.
| Table 1: | Calculated thermodynamic and other parameters for Zolpidem and its metabolites |
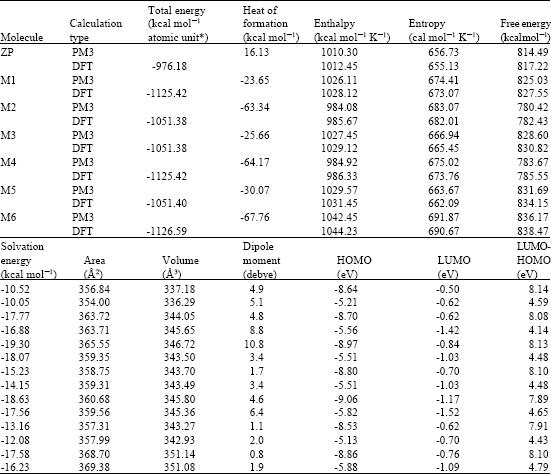 | |
| * in atomic units from DFT calculations | |
RESULTS AND DISCUSSION
Table 1 gives the total energy, heat of formation as per PM3 calculation, enthalpy, entropy, free energy, surface area, volume, dipole moment and energies of HOMO and LUMO as per both PM3 and DFT calculations for ZP and its metabolites M1, M2, M3, M4, M5 and M6. Figure 2-8 give the regions of negative electrostatic potential (greyish-white envelopes) in (a), HOMOs (where red indicates HOMOs with high electron density) in (b), LUMOs in (c) and density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) in (d) as applied to optimised structures of ZP and its metabolites M1, M2, M3, M4, M5 and M6.
The calculated solvation energies from PM3 calculations of ZP and its metabolites M1, M2, M3, M4, M5 and M6 in kcal mol-1 are respectively -10.52, -17.77, -19.30, -15.23, -18.63, -13.16 and -17.58 and corresponding dipole moments from DFT calculations are 5.1, 8.8, 3.4, 3.4, 6.4, 2.0 and 1.9. The values suggest that ZP and its metabolites would be soluble in water so that they would have a high clearance rate via urine.
The LUMO-HOMO energy differences for ZP and its metabolites from DFT calculations are found to be moderately large ranging from 4.1 to 4.8 eV, indicating that the compounds would not differ greatly in their kinetic lability and that neither would be highly labile. The metabolite M1 has the smallest LUMO-HOMO energy difference, indicating that it would be marginally more labile than the parent drug and other metabolites.
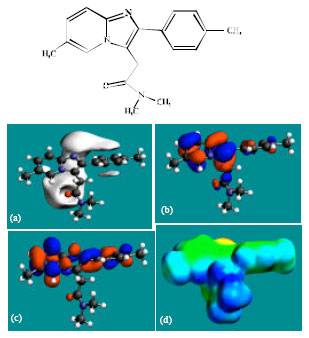 | |
| Fig. 2: | Structure of ZP giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
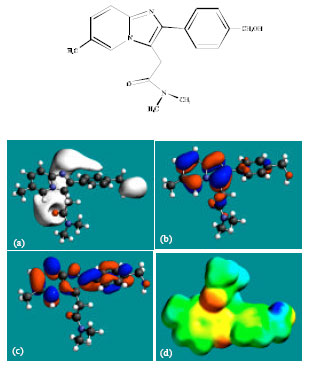 | |
| Fig. 3: | Structure of M1 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
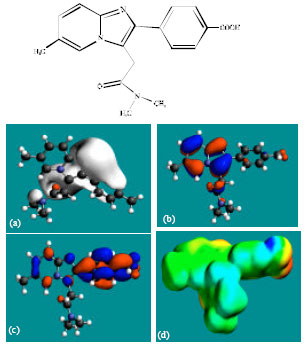 | |
| Fig. 4: | Structure of M2 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
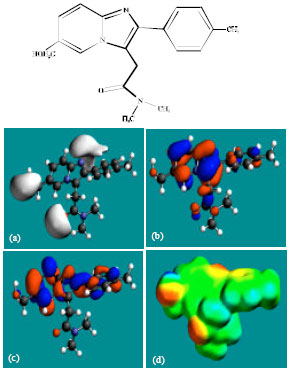 | |
| Fig. 5: | Structure of M3 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
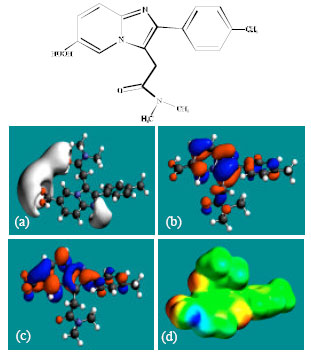 | |
| Fig. 6: | Structure of M4 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
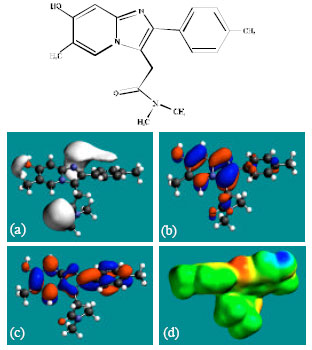 | |
| Fig. 7: | Structure of M5 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
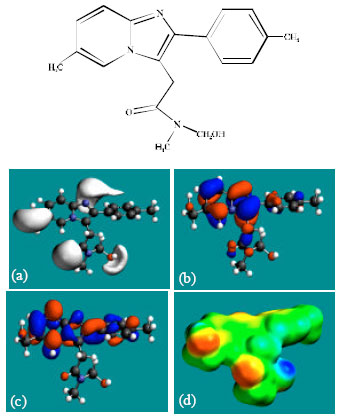 | |
| Fig. 8: | Structure of M6 giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
In the case of ZP, the electrostatic potential is found to be more negative above and below the phenyl ring and around oxygen and nitrogen centers, indicating that the positions may be subject to electrophilic attack. In the case of M1, M2, M3, M4, M5 and M6 also, the electrostatic potential is found to be more negative around oxygen and nitrogen centers, indicating that the positions may be subject to electrophilic attack.
In the case of ZP and all its metabolites M1, M2, M3, M4, M5 and M6 both the HOMOs with high electron density and the LUMOs are found to be centered mostly on the non-hydrogen atoms of phenyl, imidazole and pyridine rings.
The overlap of HOMO with high electron density and region of negative electrostatic potential at some positions, gives further support to the idea that the positions may be subject to electrophilic attack.
When the surface area and volume of ZP are compared with those of its metabolites (Table 1), it is found that the values do not differ widely so that all of the compounds may be able to act as substrate for the same binding site unless disallowed by their chemical differences.
When the heats of formation of M1 and isomeric metabolite M3 are compared (Table 1), it is found that the two values are similar, indicating that the two compounds would not differ significantly in their stability. Similarly, isomeric metabolites M2 and M4 are found to have comparable stability. However, M6 is found to be significantly more stable than isomeric metabolites M1 and M3.
The molecular surface of ZP followed by that of M4 is found to abound most in electron-deficient (blue) regions, indicating that the two compounds may be most subject to nucleophilic attacks such as those by glutathione and nucleobases in DNA. Reaction with glutathione can induce cellular toxicity by compromising the antioxidant status of the cell whereas that with nucleobases in DNA can cause DNA damage. However, as stated earlier, since ZP and its metabolites are expected to be kinetically inert, the rate of such adverse reactions may be low unless speeded up enzymatically. Since ZP and its metabolites do not differ much in their kinetic lability, greatest abundance of positively charged regions on the molecular surface would make ZP most toxic. That higher doses of ZP are associated with increased incidence of nausea and vomiting may be due to elevation in concentration of the parent drug rather than any of the metabolites, although when given in recommended doses, only a trace amount of unchanged drug has been detected only in trace amounts (Ascalone et al., 1992).
CONCLUSIONS
Zolpidem is a new orally active sleep inducer belonging to the class of compounds known as imidazopyridine. The drug is rapidly absorbed, has mean elimination half-life of about 2 h and is extensively metabolized and excreted as pharmacologically inactive metabolites. Molecular modelling analyses based on semi-empirical and DFT calculations show that ZP and its metabolites have moderately large LUMO-HOMO energy differences so that none of the compounds is expected to be highly labile. Molecular surface of ZP is found to abound in electron-deficient blue regions so that it could react with glutathione and nucleobases in DNA. However, the rates of such adverse reactions may be low because of kinetic inertness, unless these are speeded up enzymatically.
Abbreviations
| ZP: | Zolpidem; N,N,6-trimethyl-2- (4-methylphenyl) -imidazole (1,2-a) pyridine-3 -acetamide L-(+)-hemitartrate) |
| DFT: | Density functional theory |
| LUMO: | Lowest unoccupied molecular orbital |
| HOMO: | Highest occupied molecular orbital |
ACKNOWLEDGMENTS
Fazlul Huq is grateful to the Discipline of Biomedical Science, School of Medical Sciences, The University of Sydney for the time release from teaching.
REFERENCES
- Ascalone, V., L. Flaminio, P. Guinebault, J.P. Thenot and P.L. Morselli, 1992. Determination of zolpidem, a new sleep-inducing agent and its metabolites in Biological fluids: Pharmacokinetics, drug metabolism and overdosing investigations in humans. J. Chromatogr., 581: 237-250.
PubMed - Debailleul, G., F. Abi Khalil and P. Lheureux, 1991. HPLC quantification of zolpidem and prothipendyl in a voluntary intoxication. J. Anal. Toxicol., 15: 35-37.
Direct Link - Hempel, G. and G. Blaschke, 1996. Direct determination of zolpidem and its main metabolites in urine using capillary electrophoresis with laser-induced fluorescence detection. J. Chromatogr. B., 675: 131-137.
Direct Link - Langtry, H.D. and P. Benfield, 1990. Zolpidem, A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential. Drug, 40: 291-313.
PubMed - Olubodun, J.O., H.R. Ochs, L.L. Von Moltke, R. Roubenoff and L.M. Hesse et al., 2003. Pharmacokinetic properties of zolpidem in elderly and young adults: possible modulation by testosterone in men. Br. J. Clin. Pharmacol., 56: 297-304.
Direct Link - Von Moltke, L.L., D.J. Greenblatt, B.W. Granda, S.X. Duan and J.M. Grassi et al., 1999. Zolpidem metabolism in vitro: Responsible cytochromes, chemical inhibitors and in vivo correlations. Br. J. Clin. Pharmcol., 48: 89-97.
PubMed