
Neelam Seedher
Department of Chemistry, Panjab University, Chandigarh, India
Pooja Agarwal
Department of Chemistry, Panjab University, Chandigarh, India
Journal of Pharmacology and Toxicology
Year: 2006 | Volume: 1 | Issue: 1 | Page No.: 89-100







ABSTRACT
Mechanism of interaction of cefdinir with human serum albumin has been studied using fluorescence spectroscopic technique. Cefdinir showed high affinity to serum albumin with association constants of the order of 104. The percentage of drug bound decreased with increase in temperature. The effect of temperature was more significant at low drug:protein ratios. The nature of interaction was predicted from the thermodynamic parameters for the binding and studies carried out in the presence of hydrophobic probe, 1-anilinonaphthalene-8-sulfonate (ANS). The results showed that hydrophobic interactions are not involved in the binding and the interaction is predominantly through hydrogen bonding. Studies carried out in the presence of site II-specific probe, dansylsarcosine (DSS) showed that site II on HSA is involved in binding; cefdinir and DSS appear to bind at different regions within site II. Further it was found that at pH 8, cefdinir causes conformational change in the albumin molecule which resulted in increase in the binding constants and consequent decrease in the fraction of free pharmacologically active drug. The presence of salt (0.15 M NaCl), on the other hand, was associated with significant reduction in the extent of binding and increase in the percentage of free drug. The Stern-Volmer analysis of the binding data indicated that the tryptophan residues of albumin are not fully accessible to the drug and predominantly static quenching mechanism is operative.
PDF Abstract XML References
How to cite this article
Neelam Seedher and Pooja Agarwal, 2006. Mechanism of Interaction of Anti-microbial Drug Cefdinir with Human Serum Albumin. Journal of Pharmacology and Toxicology, 1: 89-100.
DOI: 10.3923/jpt.2006.89.100
URL: https://scialert.net/abstract/?doi=jpt.2006.89.100
DOI: 10.3923/jpt.2006.89.100
URL: https://scialert.net/abstract/?doi=jpt.2006.89.100
INTRODUCTION
Cefdinir (FK482), an extended-spectrum third-generation semisynthetic cephalosporin, exhibits an excellent activity against a wide range of gram-positive and gram-negative bacteria. Cefdinir inhibits the third and final stage of bacterial cell wall synthesis by preferentially binding to specific penicillin-binding proteins. Because of its hydroxyamino functionality, cefdinir is resistant to hydrolysis by 13 of the common beta-lactamases (Guay, 2000, 2002). It is rapidly absorbed from the gastrointestinal tract and is almost entirely eliminated via renal clearance of unchanged drug. cefdinir is approved for treatment of skin infections and a variety of upper and lower respiratory tract infections in both adult and pediatric patients (Perry and Scott, 2004).
The mechanism of binding of drugs to plasma proteins has important pharmacokinetic and pharmacodynamic implications since only the free drug is pharmacologically active (Kragh-Hansen et al., 2002). Binding of some cephalosporins to human serum albumin has been reported (Fernandez et al., 1993; Terasaki et al., 1992; Nerli et al., 1997). Structural specificity requirements, nature of interaction, correlation of binding affinity with lipophilicity and the interaction between cefotaxime and serum albumin of several mammalian species have been studied. However, such studies on cefdinir are not available. Although cephalosporins are structurally and pharmacologically related, they are bound to serum albumin with diverse affinity depending upon their side chain structure and must be considered individually, generalizations cannot be made (Briand et al., 1982). In the present study, detailed molecular mechanism of interaction of cefdinir with human serum albumin has been studied using fluorescence spectroscopic technique. Binding parameters, thermodynamics of the binding process, fluorescence quenching mechanism, the nature of forces involved in the interaction and the effect of pH and presence of salt on the biologically active free drug have been reported. Such studies are useful since free rather than total drug concentrations are more reliable parameters for the therapeutic monitoring of drugs (Greenblatt et al., 1982).
MATERIALS AND METHODS
Materials
Pure cefdinir drug sample was obtained as gift from Aurobindo Pharma Ltd., Hyderabad, India. Human Serum Albumin (HSA), fuorescent probes, 1-anilinonaphthalene-8-sulfonate (ANS) and dansylsarcosine piperidinium salt (DSS) were purchased from Sigma Chemical Co., USA. All other reagents were of analytical grade. Water used was double distilled in all glass apparatus. HSA solutions were prepared based on molecular weight of 66,500. All experiments were carried out in 0.1M phosphate buffer using fluorescence spectroscopic technique. Perkin Elmer fluorescence spectrophotometer (MPF 44B) equipped with a 150 W xenon lamp source was used.
Methods
Determination of Binding Parameters
For the determination of binding parameters, two millilitres of 10 μM albumin solution was taken in a quartz cell and increasing amounts of drug stock solution (400 μM) was added. The final drug concentration was in the range 5-60 μM. Albumin concentration was kept fixed at 10 μM by adding the same volume of 20 μM albumin to the cell. Fluorescence spectra were recorded in the range 300-400 nm after excitation at 295nm, in each case. Intrinsic fluorescence of protein was measured at 332 nm, drug used did not have any fluorescence at the emission wavelength of protein. The fluorescence data was corrected for inner filter effect (Oberfelder and Lee, 1985) using equation
| (1) |
where, Fcorr and Fobs are the corrected and observed fluorescence intensity and ODex and ODem are the optical density of the sample at the excitation and emission wavelengths, respectively.
Stoichiometry of the drug-protein interaction was determined by the method of continuous variations (Job 1928; Rahman et al., 1993) The fluorescence change (ΔF = Fprotein-Fprotein + drug ) of a series of protein-drug mixtures was measured under such conditions that the total concentration of drug plus protein was held constant at 10 μM but the respective mole fraction of each was varied. ΔF was plotted against the mole fraction of drug (Job’s plot) and the stoichiometry of binding was obtained from the maximum in the plot in each case.
Data Analysis
Data was analysed as follows using Ward (1985) method. The fractional occupancy of the total protein binding sites by drug was obtained from the ratio, θ = ΔF/ΔFmax (Weber and Young 1964; Maruyama et al., 1990) where, ΔF = F0 – F, F0 and F are the fluorescence intensities of serum albumin in the absence and presence of drug, respectively. ΔFmax values were obtained from the double reciprocal (1/ΔF versus 1/ Dt) plots.
If Pt is the total protein concentration and n is the number of binding sites, the total number of sites on protein is given by nPt and the concentration of bound sites on protein is given by nθPt (Ward, 1985) which is also equal to the concentration of the bound drug (Db). Df, the number of moles of free drug, was obtained from the difference, Dt - Db, where Dt is the total drug added. The amount bound was expressed as moles of drug bound per mole protein, r (=Db/Pt). The binding parameters were computed directly by fitting the experimental data (r and Df values) to the following general equation (Scatchard equation) using an iterative non-linear least squares regression program developed for this purpose.
| (2) |
The association constant (Ka) and the number of binding sites (n) were determined at three different temperatures (17, 27 and 37°C) and the thermodynamic parameters for binding were calculated using equations
| (3) |
| (4) |
The reported data are an average of three determinations with coefficient of variation less than 2% in each case. The percentage of drug bound (β = Db/Dtx100) was calculated from the association constants using the relationship, β = {[Pt]/([Pt] + (1/Ka) + [Df])}x100 (Martin 1965).
To study the effect of pH on binding parameters, the temperature was kept at 27°C and the experiments were conducted at three different pH values; 6.4, 7.4 and 8.0. The effect of the presence of salt was studied at 27°C and pH 7.4 by using phosphate buffer saline (containing 0.15 M NaCl as the electrolyte). The percentage of free drug (α = Df/Dtx100) was calculated from the dissociation constants for drug-protein complex (Kd = 1/Ka) using the relationship (Kd + Df)/{[Pt] + Kd + [Df]}x100 (Martin, 1965).
Drug-albumin Interaction in the Presence of Fluorescent Probes
Hydrophobic Probe, ANS
Experiments were also carried out in the presence of hydrophobic probe, ANS (Daniel and Weber 1966; Seedher et al., 1999). HSA-ANS interaction was studied in the presence and absence of drug. Increasing amounts of drug was added to an equimolar albumin-ANS mixture (10 μM each). The drug concentration was varied from 1 to 12 μM and the concentration of HSA-ANS mixture was kept fixed at 10 μM each by adding the same volume of HSA-ANS mixture (20 μM each) to the cell. The fluorescence of ANS was measured at 470 nm after excitation at 370 nm.
Site-selective Probe
Fluorescence probe displacement experiments were carried out using site II-selective probe, Dansylsarcosine (DSS) (Sudlow et al., 1975, 1976). The fluorescence of the DSS was measured at 27°C in DSS-HSA mixture (1:1, 5 μM each) before and after the addition of drug (2-28 μM). DSS fluorescence was measured at 480 nm after excitation at 350 nm. The percentage displacement of probe was determined using equation (F1-F2)/F1, Where, F1 and F2 are the fluorescence intensities of the probe + HSA without and with drug, respectively. The probe to HSA ratio was kept 1:1 in order to keep the non-specific binding of probe to a minimum.
RESULTS
Drug-serum Albumin Interaction
The structure of cefdinir is shown in Fig. 1. The drug was found to quench the intrinsic fluorescence of human serum albumin. However, there was no significant shift in the wavelength for maximum emission (332 nm) in all cases except at pH 8.0, indicating thereby that under these conditions the binding does not cause any major conformational change in the protein molecule. At pH 8.0, a 10 nm blue shift in the fluorescence emission spectrum was observed. The stoichiometry of the interaction was determined by the method of continuous variations, described in the experimental section. The maximum in the fluorescence change in the Job’s plot occurred at 0.5 mole fraction of drug corresponding to 1:1 stoichiometry (Fig. 2).
The association constants for the binding and the number of binding sites, determined by the non-linear least squares regression program, are given in Table 1. The experimental data could be fitted into an equation for only one class of binding sites (m = 1). Association constants were of the order of 104. Percentage of drug bound at different temperatures is plotted in Fig. 3 against total drug added per mol protein (Dt/Pt).
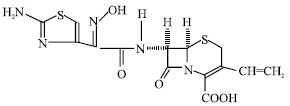 | |
| Fig. 1: | Structure of cefdinir |
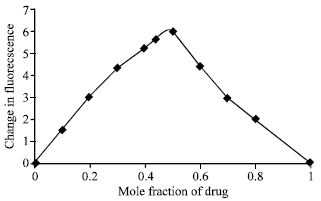 | |
| Fig. 2: | Job’s plot for HSA-cefdinir system |
| Table 1: | Binding parameters for the interaction of cefdinir with human serum albumin at pH 7.4 and different temperatures |
 | |
| The binding parameters were computed directly by fitting the experimental data (r and Df values) to the Scatchard equation using an iterative non-linear least squares regression program | |
| Table 2: | Thermodynamic parameters for the interaction of cefdinir with human serum albumin |
 | |
| * ΔG° values have been calculated at 37°C | |
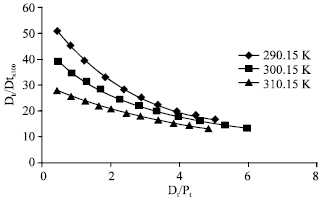 | |
| Fig. 3: | Percentage of drug bound at different temperatures |
With increase in temperature the percentage of drug bound decreased, the effect being more significant at low drug:protein ratios. The association constants also decreased with increase in temperature while the number of binding sites remained almost same. Thermodynamic parameters for the binding, determined from the temperature-dependence of the association constants, are given in Table 2. Free energy change (ΔG°) was negative. Both ΔH° and ΔS° were also found to have high negative values.
Drug-albumin Interaction in the Presence of Fluorescent Probes
In order to further understand the nature of interaction involved, binding with HSA was also studied in the presence of hydrophobic probe, ANS (Daniel and Weber 1966; Seedher and Bhatia, 2005) and site II-specific probe, dansylsarcosine (DSS) (Sudlow et al., 1975, 1976). ANS fluorescence was measured in HSA-ANS system. Addition of drug to HSA-ANS system did not cause any significant change in the fluorescence of ANS. The addition of increasing amount of drug to HSA-DSS mixture resulted in the decrease of DSS fluorescence. The percentage displacement was about 31% at the highest drug concentration used (28 μM). Percentage displacement (D) has also been plotted against drug concentration in Fig. 4. The maximum displacement, obtained from the double reciprocal plot (1/D versus 1/Dt), was found to be 64.5%.
| Table 3: | Effect of pH and presence of salt on the binding parameters of cefdinir with human serum albumin at 27°C |
 | |
| The binding parameters were computed directly by fitting the experimental data (r and Df values) to the Scatchard equation using an iterative non-linear least squares regression program | |
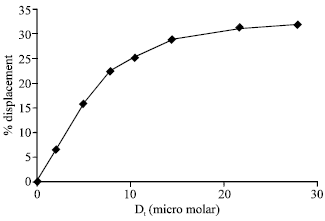 | |
| Fig. 4: | Percentage displacement of dansylsarcosine from HSA by cefdinir |
Effect of pH and the Presence of Salt
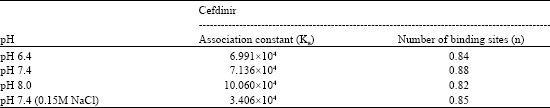
Association constants for the binding and the number of binding sites at three different pH values, 6.4, 7.4 and 8.0, determined at 27°C are given in Table 3. Increase of pH from 6.4 to 7.4 did not cause any significant change in the association constants but the value increased significantly on increasing the pH to 8.0. The number of binding sites remained almost same at all pH values. Association constant significantly decreased in the presence of 0.15M NaCl. Percentage of free drug at pH 7.4, pH 8.0 and in the presence of 0.15M NaCl is plotted in Fig. 5 against total drug added per mol protein (Dt/Pt). The percentage of free drug decreased with increase in pH from 7.4 to 8.0, the effect being more significant at low drug:protein ratios. A very significant increase in the percentage of free drug was observed in the presence of salt (0.15M NaCl).
Stern-volmer Analysis
Fluorescence quenching data could not be fitted to the simple Stern-Volmer equation (Eftink and Ghiron, 1981)
| (5) |
| Table 4: | Stern-Volmer parameters, Kq and fa for the interaction of cefdinir with human serum albumin at different temperatures |
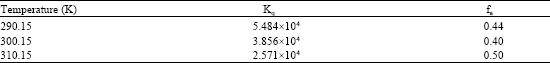 | |
| (6) |
The results were, therefore, analysed by the modified Stern-Volmer equation (Lehrer 1971, Seedher and Kanojia, 2001) where F0 and F are the fluorescence intensities at 332 nm in the absence and presence of quencher (drug), respectively, Kq is the Stern-Volmer quenching constant and fa is the fraction of fluorophore (protein) accessible to the quencher (drug). From a plot of F0/(F0 - F) versus 1/ DT, fa and Kq were determined. Kq values were of the order of 104 and were found to decrease with increase in temperature and fa values were found to lie between 0.4-0.5. Kq and fa values at different temperatures are given in Table 4.
DISCUSSION
Cefdinir is an anionic drug at physiologic pH. It has three ionisable groups, carboxyl, amino and hydroxyl with pKa values 1.9, 3.3 and 9.9, respectively (Lepsy et al., 2003). Cefdinir was found to exhibit strong binding to albumin with only one class of binding sites. Nerli et al. (1997) studying the interaction of a large number of cephalosporins with HSA has shown that the association constants can vary over a very wide range. Nerli et al. (1997) has classified cephalosporins into three groups according to their affinity for albumin: low affinity (K = 10-102 M-1), medium affinity (K = 103 M-1) and high affinity (K = 104 M-1). They have also reported that the presence of rich electron density substituents at R1 and R2 positions increase the affinity of drugs for albumin. According to this classification, cefdinir with high electron density substituent at R1 position falls into the high affinity category with association constants (Ka) of the order of 104. Percentage of drug bound decreased with increase in temperature and with increase in the drug:protein molar ratio (Fig. 3). At low drug:protein ratios, a significant fraction of the added drug was bound in each case. It may be mentioned that low drug:protein ratios are frequently encountered in the physiological system since in blood, the serum albumin concentration is very large (0.53-0.75 mM).
The nature of drug-protein interaction could be predicted from the thermodynamic parameters for the binding. For electrostatic interactions, ΔH0 is very small, nearly zero (Ross and Subramanian, 1981; Aki and Yamamoto, 1989) and thus electrostatic interactions are not present. Negative ΔS0 values indicate that hydrophobic interactions are not involved since hydrophobic interactions lead to increase in entropy of the system (Miyoshi et al., 1992). Negative enthalpy and entropy changes arise from van der Waals’ interactions and hydrogen bond formation (Plaizier and De Neve, 1982). High negative ΔH° values, however, indicate that in the present case, the drug-protein interaction is predominantly through hydrogen bonding (Seedher et al., 1999). In the literature a diverse kind of binding forces have been reported for the binding of cephalosporins to albumin. Fernandez et al. (1993) have shown the existence of ionic and hydrogen bonds, Briand et al. (1982) have shown the interactions to be principally electrostatic. However, information on the nature of interaction of cefdinir with human serum albumin is not available since cefdinir has not been included in the previous studies.
It is known that the hydrophobic probe, ANS shows greatly increased fluorescence as a result of hydrophobic interaction with proteins and other macromolecules due to the transfer of the probe from an aqueous to a non-polar environment (Jun and Ruenitz, 1978). No significant change in ANS fluorescence on the addition of drug to HSA-ANS system shows that drug does not displace ANS from its binding site. Thus ANS and drug do not share a common site on the albumin molecule. Since ANS binds predominantly to hydrophobic surface, it may again be concluded that hydrophobic interactions are not involved in the binding of cefdinir to HSA.
Site-specific probe, dansylsarcosine (DSS) also shows greatly enhanced fluorescence as a result of interaction with proteins and other macromolecules (Sudlow et al., 1975; Sudlow et al., 1976). The addition of increasing amount of drug to HSA-DSS mixture resulted in decrease of DSS fluorescence, indicating thereby that cefdinir displaces DSS from its binding site. Since DSS is a site II-specific probe, displacement of DSS from its binding site on albumin showed that site II is involved in the binding of cefdinir to HSA. However, the maximum displacement, obtained from the double reciprocal plot was only 64%, indicating that cefdinir and DSS bind at different regions within site II. Crystallographic analyses have assigned site II in HSA to subdomain IIIA and among the individual amino acid residues in this subdomain, 410Arg and 411Tyr are usually assumed to be important (Sugio et al., 1999; He and Carter 1992). Nerli et al. (1997) have also shown that site II possesses capacity to bind a large number of cephalosporins.
Increase of pH from 6.4 to 7.4 did not cause any significant change in the association constants but the value increased significantly on increasing the pH to 8.0. The variation in the magnitude of binding parameters can either be due to the change in the degree of ionization of the drug (or protein) or due to conformational changes in the protein molecule. The drug cefdinir contains three ionizable groups, carboxyl, amino and hydroxyl with pKa values 1.9, 3.3 and 9.9, respectively (Lepsy et al., 2003). No significant change in the ionization of these groups is expected in the pH range 6.4 to 8.0. Carboxyl groups will remain almost fully ionized while amino and hydroxyl groups will remain almost fully unionized in this pH range. In the albumin molecule with increase in pH, the fraction of cationic surface decreases while anionic and neutral surface increases (Carter and Ho, 1994) and this also could not explain the significant increase in binding constant at pH 8.0. The results could only be explained on the basis of conformational flexibility in the albumin molecule. HSA molecule is known to undergo pH-dependant conformational transitions but these generally occur below pH 4.5 and above pH 8.0 (Carter and Ho, 1994). It thus appears that the presence of cefdinir causes a conformational change in the albumin molecule as the pH is increased to 8.0 and this may be responsible for the increase in the binding constant at this pH. This explanation found support from the observation that while at pH 6.4 and 7.4 there was no shift in the wavelength for maximum emission (λmax) of HSA; at pH 8.0 the addition of drug caused a 10 nm decrease in λmax. The large blue shift in λmax appears to be due to the conformational change in the albumin molecule caused by drug at pH 8.0. The increase in the extent of binding results in the reduction of free fraction (Fig. 5). Further studies are needed to explore the secondary structural changes caused by cefdinir at alkaline pH in the albumin molecule.
The presence of salt decreased the association constants but the number of binding sites remained almost same. Since thermodynamic parameters for the binding of these drugs do not suggest electrostatic interactions, it appears that chloride ions displace the drug from its binding site and hence the binding constants are lowered in the presence of salt. Similar findings have also been reported by Afifi (1999), Wilting et al. (1981) using warfarin and some non-steroidal anti-inflammatory drugs. It has been shown by Wilting et al. (1981) that chloride ions also affect N-B transition but at pH 7.4 a competition between drug and chloride ions is dominant. The fact that chloride ions also bind to site II of HSA and chloride ions displace drugs from their binding site, confirms the conclusion that drug is bound at site II on HSA.
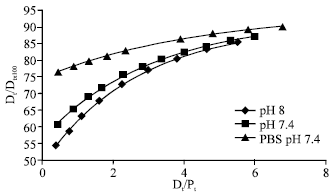 | |
| Fig. 5: | Percentage of drug free at different pH values and in the presence of salt |
The reduction in the extent of binding resulted in significant increase in the concentration of free pharmacologically active drug in the presence of salt (Fig. 5).
It is known that excitation at 295 nm involves fluorescence due to tryptophan residues of protein (Williams et al., 1965). Stern-Volmer analysis of fluorescence data is useful in the estimation of the accessibility of tryptophan residues in proteins to the drug (quencher) molecules and in understanding the quenching mechanism. Since simple Stern-Volmer equation could not be fitted to the data, it implies that all the tryptophan residues are not fully accessible to the drug molecules. It has also been reported by Williams et al. (1965) that under conditions of the experiment, since tryptophan residues are not fully exposed, accessibility of interacting species varies with the nature and size of its molecules. However, the data could be fitted to modified Stern-Volmer equation which takes into account the fraction of fluorophore accessible to the quencher (fa). fa values calculated from the modified Stern-Volmer plots showed that only a fraction of tryptophan residues are accessible to drug. Decrease in the quenching constant with increase in temperature and the observation that the quenching constants are close to the association constants for the interaction indicated that the quenching mechanism essentially involves static quenching (Gonzalez-Jimenez et al., 1992).
For a bimolecular quenching process, Kq = kq τ0 where τ0 is the lifetime in the absence of quencher and kq is the rate constant for quenching. As τ0 value for tryptophan fluoresence in proteins is known to be of the order of 10-9s (Cui et al., 2004), the rate constant, kq would be of the order of 1013 M-1s-1. The upper limit of kq expected for a diffusion-controlled bimolecular collisional quenching constant is 1010 M-1s-1 (Pang et al., 2005). The high magnitude of kq in the present study (1013 M-1s-1) also shows that the quenching is not initiated by dynamic collision, but originates from the formation of a complex. However, collisional quenching mechanism is also involved since the magnitude of quenching constants are smaller than the association constants for the interaction.
CONCLUSIONS
Cefdinir exhibits strong binding to human serum albumin with only one class of binding sites. Binding constants were of the order of 104. Thermodynamic parameters for the binding and the studies carried out in the presence of hydrophobic probe showed that hydrophobic interactions are not involved, the binding is predominantly through hydrogen bonding interactions. Fluorescence probe displacement studies showed that site II of human serum albumin is involved in the interaction of cefdinir with HSA. Cefdinir causes a conformational change in the albumin molecule at pH 8, resulting in increase in the binding constant and decrease in the free pharmacologically active drug. The presence of salt resulted in decrease in the binding constants and significant increase in the concentration of free drug. Stern-Volmer analysis of the fluorescence data showed that the quenching mechanism essentially involves static quenching and the tryptophan residues of albumin are not fully accessible to drug molecules.
ACKNOWLEDGMENTS
One of the authors, Pooja Agarwal is thankful to the council of scientific and industrial research (CSIR), New Delhi, India for financial assistance. The authors are also thankful to M/s. Aurobindo Pharma Ltd., Hyderabad, India for the gift sample of cefdinir.
REFERENCES
- Afifi, N.N., 1999. Using difference spectrophotometry to study the influence of different ions and buffer systems on drug protein binding. Drug Dev. Indus. Pharm., 25: 735-743.
Direct Link - Aki, H. and M. Yamamoto, 1989. Thermodynamics of the binding of phenothiazines to human plasma, human serum albumin and alpha-acid glycoprotein: A calorimetric study. J. Pharma. Pharmacol., 41: 674-679.
PubMedDirect Link - Briand, C., M. Sarrazin, V. Peyrot, R. Gilli, M. Bourdeaux and J.C. Sari, 1982. Study of interaction between human serum albumin and some cephalosporins. Mol. Pharmacol., 21: 92-99.
Direct Link - Carter, D.C. and J.X. Ho, 1994. Structure of serum albumin. Adv. Protein Chem., 45: 153-203.
CrossRefPubMedDirect Link - Craig, W.A. and C.M. Kunin, 1976. Significance of serum protein and tissue binding of antimicrobial agents. Ann. Rev. Med., 27: 287-300.
CrossRefDirect Link - Cui, F.L., J. Fan, W. Li, Y.C. Fan and Z.D. Hu, 2004. Fluorescence Spectroscopic studies on 5-aminosalicylic acid and zinc 5-aminosalicylate interaction with human serum albumin. J. Pharm. Biomed. Anal., 34: 189-197.
Direct Link - Eftink, M.R. and C.A. Ghiron, 1981. Fluorescence quenching studies with proteins. Anal. Biochem., 114: 199-227.
PubMedDirect Link - Fernandez, G.M., J.M. Lumbreras and D. Ordonez, 1993. A thermodynamic approach to the binding mechanisms of cefotaxime to serum albumins. J. Pharm. Sci., 82: 948-951.
PubMedDirect Link - Greenblatt, D.J., E.M. Sellers and J. Koch-Weser, 1982. Importance of protein binding for the interpretation of serum or plasma drug concentrations. J. Clin. Pharmacol., 22: 259-263.
Direct Link - Jimenez, J.G., G. Frutos and I. Cayre, 1992. Fluorescence quenching of human serum albumin by xanthines. Biochem. Pharmacol., 44: 824-826.
Direct Link - Guay, D.R., 2000. Cefdinir: An expanded-spectrum oral cephalosporin. Ann. Pharmacother., 34: 469-477.
CrossRefDirect Link - Guay, D.R., 2002. Cefdinir: An advanced-generation, broad-spectrum oral cephalosporin. Clin. Ther., 24: 473-489.
CrossRefDirect Link - He, X.M. and D.C. Carter, 1992. Atomic structure and chemistry of human serum albumin. Nature, 358: 209-215.
CrossRefDirect Link - Jun, H.W. and P.C. Ruenitz, 1978. Interaction of tricyclic antipsychotic and antidepressant drugs with 1-anilino-8-naphthalenesulfonic acid. J. Pharm. Sci., 67: 861-863.
PubMedDirect Link - Lehrer, S.S., 1971. Solute perturbation of protein fluorescence. The quenching of tryptophan fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry, 10: 3254-3263.
CrossRefDirect Link - Lepsy, C.S., R.J. Guttendorf, A.R. Kugler and D.E. Smith, 2003. Effects of organic anion, organic cation and dipeptide transport inhibitors on cefdinir in the isolated perfused rat kidney. Antimicrob. Agents Chemother., 47: 689-696.
CrossRefPubMedDirect Link - Lin, J.H., D.M. Cocchetto and D.E. Duggan, 1987. Protein binding as a primary determinant of the clinical pharmacokinetic properties of non-steroidal anti-inflammatory drugs. Clin. Pharmacokinet, 12: 402-432.
PubMedDirect Link - Martin, B.K., 1965. Potential effect of the plasma proteins on drug distribution. Nature, 207: 274-276.
CrossRefDirect Link - Miyoshi, T., K. Sukimoto and M. Otagiri, 1992. Investigation of the interaction mode of phenothiazine neuroleptics with alpha 1-acid glycoprotein. J. Pharm. Pharmacol., 44: 28-33.
PubMedDirect Link - Nerli, B., D. Romanini and G. Pico, 1997. Structural specificity requirements in the binding of beta lactam antibiotics to human serum albumin. Chemico-Biol. Interact., 104: 179-202.
PubMedDirect Link - Oberfelder, R.W. and J.C. Lee, 1985. Measurement of ligand-protein interaction by electrophoretic and spectroscopic techniques. Methods Enzymol., 117: 381-388.
PubMedDirect Link - Pang, Y.H., L.L. Yang, S.M. Shuang, C. Dong and M. Thompson, 2005. Interaction of human serum albumin with bendroflumethiazide studied by fluorescence spectroscopy. J. Photochem. Photobiol. B: Biol., 80: 139-144.
PubMedDirect Link - Perry, C.M. and L.J. Scott, 2004. Cefdinir: A review of its use in the management of mild-to-moderate bacterial infections. Drugs, 64: 1433-1464.
PubMedDirect Link - Plaizier-Vercammen, J.E. and R.E. De Neve, 1982. Interaction of povidone with aromatic compounds III: Thermodynamics of the binding equilibria and interaction forces in buffer solutions at varying pH values and varying dielectric constant. J. Pharm., Sci., 71: 552-556.
PubMedDirect Link - Rahman, M.H., T. Maruyama, T. Okada, K. Yamasaki and M. Otagiri, 1993. Study of interaction of carprofen and its enantiomers with human serum albumin-I. Mechanism of binding studied by dialysis and spectroscopic methods. Biochem. Pharmacol., 46: 1721-1731.
Direct Link - Ross, P.D. and S. Subramanian, 1981. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry, 20: 3096-3102.
CrossRefDirect Link - Seedher, N. and S. Bhatia, 2005. Mechanism of interaction of non-steroidal antiiflammatory drugs meloxicam and nimesulide with serum albumin. J. Pharm. Biomed. Anal., 39: 257-262.
Direct Link - Seedher, N. and M. Kanojia, 2001. Mechanism of interaction of phenothiazine derivatives with serum albumin. Indian J. Pharm. Sci., 63: 137-143.
Direct Link - Seedher, N., B. Singh and P. Singh, 1999. Mode of interaction of metronidazole with bovine serum albumin. Indian J. Pharm. Sci., 61: 143-148.
Direct Link - Sudlow, G., D. Birkett and D. Wade, 1975. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol., 11: 824-832.
PubMedDirect Link - Sudlow, G., D. Birkett and D. Wade, 1976. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol., 12: 1052-1061.
Direct Link - Sugio, S., A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, 1999. Crystal structure of human serum albumin at 2.5 a resolution. Protein Eng., 12: 439-446.
PubMedDirect Link - Terasaki, T., H. Nouda and A. Tsuji, 1992. Relation between lipophilicity and binding affinity with HSA for penicillin and cephem antibiotics. J. Pharmacobio-Dyn, 15: 99-106.
PubMedDirect Link - Weber, G. and L.B. Young, 1964. Fragmentation of bovine serum albumin by pepsin. I. The origin of the acid expansion of the albumin molecule. J. Biol. Chem., 239: 1415-1423.
PubMedDirect Link - Williams, E.J., T.T. Herskovits and M. Laskowski, 1965. Location of chromophoric residues in proteins by solvent perturbation. 3. Tryptophyls in lysozyme and in α-chymotrypsinogen and its derivatives. J. Biol. Chem., 240: 3574-3579.
PubMedDirect Link