
Fazlul Huq
School of Biomedical Sciences, Faculty of Health Sciences,
The University of Sydney, Australia
Journal of Pharmacology and Toxicology
Year: 2006 | Volume: 1 | Issue: 4 | Page No.: 362-368







ABSTRACT
Adefovir dipivoxil (ADV) is an oral prodrug designed to enhance low intestinal absorption of the anti-viral agent adepovil (PMEA). The pivoxil moieties of ADV are rapidly cleaved to produce PMEA during absorption through the gut wall. After entry into the cell, PMEA is phosphorylated to produce ADMP which is further phosphorylated to form ADDP. The antiviral activity of the drug is based on the capacity of ADDP to preferentially inhibit viral DNA replication with relative sparing of host DNA synthesis. The dose-limiting toxicity of ADV is nephrotoxicity associated with high systemic exposure. Molecular modelling analyses based on molecular mechanics, semi-empirical (PM3) and DFT (at B3LYP/6-31G* level) calculations show that ADV and its three metabolites PMEAA, ADMP and ADDP have large LUMO-HOMO energy differences so that they all would be kinetically inert. Thus, although the molecules have some electron-deficient regions on their surface so that they could potentially react with glutathione and nucleobases in DNA, the high kinetic inertness of the molecules is believed to provide protection against such adverse reactions.
PDF Abstract XML References
How to cite this article
Fazlul Huq, 2006. Molecular Modelling Analysis of the Metabolism of Adefovir Dipivoxil. Journal of Pharmacology and Toxicology, 1: 362-368.
DOI: 10.3923/jpt.2006.362.368
URL: https://scialert.net/abstract/?doi=jpt.2006.362.368
DOI: 10.3923/jpt.2006.362.368
URL: https://scialert.net/abstract/?doi=jpt.2006.362.368
INTRODUCTION
Adefovir dipivoxil [ADV, bis(pivaloyloxymethyl)-9-(2-phosphonoylmethoxyethyl)adenine] is an oral prodrug designed to enhance low intestinal absorption of the anti-viral agent adepovil [PMEA, 9-[2-(phosphonomethoxy)ethyl]adenine] which is a reverse transcriptase inhibitor (Starret et al., 1994). ADV has shown to be effective against hepatitis B virus (HBV) in both HBV e antigen-positive and HBV e antigen-negative patients (Marcellin et al., 2003, Hadziyannis et al., 2003). Chronic hepatitis B remains a global public health problem despite the availability of an effective vaccine (Raney et al., 2003). The World Health Organization estimates that approximately 400 million people worldwide are chronically infected with HBV of which at least 30% will develop cirrhosis and/or hepatocellular carcinoma (Ray et al., 2004). Also, HBV recurrence and de novo HBV are frequent events in liver transplantation recipients (Barcena et al., 2005).
The pivoxil moieties of ADV that serve to enhance its cellular permeability (Srinivas et al., 1993) are rapidly cleaved, both chemically and by esterase activity, to produce PMEA during absorption through the gut wall (Naesens et al., 1996). After entry into the cell, PMEA is phosphorylated to produce adefovir monophosphate (ADMP) by adenylate kinase 2 (Robbins et al., 1995). ADMP is further phosphorylated to form adefovil diphosphate (ADDP). The antiviral activity of PMEA is based on the capacity of ADDP to preferentially inhibit viral DNA replication with relative sparing of host DNA synthesis (Balzarini et al., 1991; De Clercq, 1993).
The dose-limiting toxicity of ADV is nephrotoxicity associated with high systemic exposure (Fisher et al., 1999). In this study, molecular modelling analyses have been carried out using the program Spartan ’04 (Spartan 2004) to investigate the relative stability of ADV and its metabolites with the aim of providing a better understanding on their relative toxicity.
Computational Methods
The geometries of ADV and its metabolites have been optimized based on molecular mechanics, (Fig. 1) semi-empirical and DFT calculations, using the molecular modelling program Spartan ’04. Molecular mechanics calculations were carried out using MMFF force field. Semi-empirical calculations were carried out using the routine PM3. DFT calculations were carried at B3LYP/6-31G* level. In optimization calculations, a RMS gradient of 0.001 was set as the terminating condition. For the optimized structures, single point calculations were carried out to give heat of formation, enthalpy, entropy, free energy, dipole moment, solvation energy, energies for HOMO and LUMO. The order of calculations: Molecular mechanics followed by semi-empirical followed by DFT ensured that the structure was not embedded in a local minimum. To further check whether the global minimum was reached, some calculations were carried out with improvable structures. It was found that when the stated order was followed, structure corresponding to the global minimum or close to that could ultimately be reached in all cases.
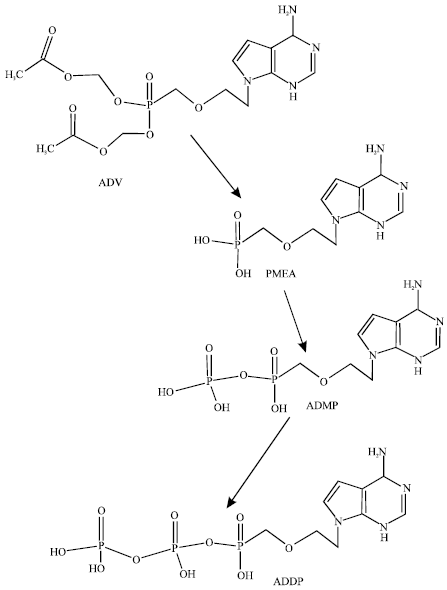 | |
| Fig. 1: | Metabolic pathways for ADV (Ray et al., 2004) |
Although RMS gradient of 0.001 may not be sufficiently low for vibrational analysis, it is believed to be sufficient for calculations associated with electronic energy levels.
RESULTS AND DISCUSSION
Table 1 gives the total energy, heat of formation as per PM3 calculation, enthalpy, entropy, free energy, surface area, volume, dipole moment, energies of HOMO and LUMO as per both PM3 and DFT calculations for ADV and its metabolites PMEA, ADMP and ADDP. Figure 2-5 give the regions of negative electrostatic potential (greyish-white envelopes) in (a), HOMOs (where red indicates HOMOs with high electron density) in (b), LUMOs in (c) and density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) in (d) as applied to the optimized structures of ADV and its metabolites PMEA, ADMP and ADDP.
The calculated dipole moments from DFT calculations of ADV and its metabolites PMEA, ADMP and ADDP are, respectively 6.9, 1.5, 4.7 and 5.3.
In the case of ADV, PMEA, ADMP and ADDP, the electrostatic potential is found to be more negative around the various oxygen and nitrogen centers, indicating that the positions may be subject to electrophilic attack.
In the case of PMEA, ADMP and ADDP both the HOMOs with high electron density and the LUMOs are found close to the non-hydrogen atoms of the six-membered heterocyclic ring. The overlap of HOMO with high electron density and region of negative electrostatic potential close to sulfur, gives further support to the idea that the position may be subject to electrophilic attack.
The molecular surfaces of ADV, PMEA, ADMP and ADDV, are found to have electron-deficient (blue), neutral (green) and negative (yellow and red) regions indicating that the compounds may be subject to nucleophilic, hydrophobic and electrophilic attacks. Within the cell, the nucleophilic attack may be that due to glutathione and nucleobases in DNA. Reaction with glutathione will induce cellular toxicity by compromising the antioxidant status of the cell whereas that with nucleobases in DNA will cause DNA damage. However, as stated earlier, since ADV and all its metabolites are expected to be kinetically inert, the rate of such adverse reactions may be low.
| Table 1: | Calculated thermodynamic and other parameters of ADV and its metabolites |
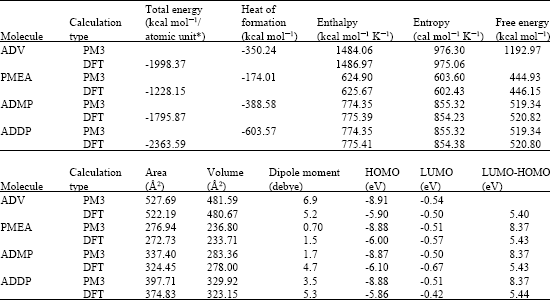 | |
| * in atomic units from DFT calculations | |
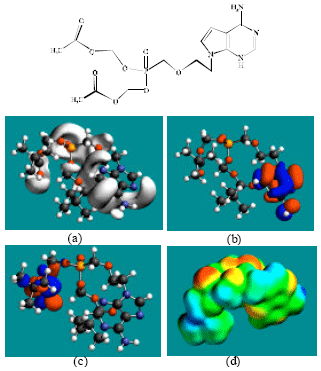 | |
| Fig. 2: | Structure of ADV giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
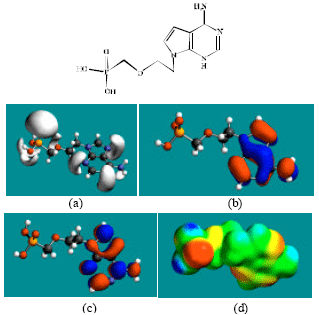 | |
| Fig. 3: | Structure of PMEA giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
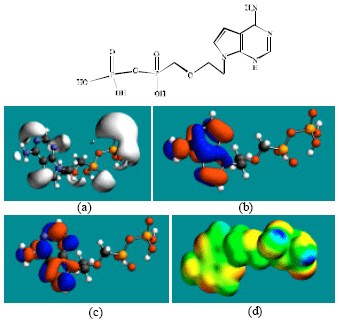 | |
| Fig. 4: | Structure of ADMP giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
 | |
| Fig. 5: | Structure of ADDP giving in: (a) the electrostatic potential (greyish envelope denotes negative electrostatic potential), (b) the HOMOs, (where red indicates HOMOs with high electron density) (c) the LUMOs (where blue indicates LUMOs) and in (d) density of electrostatic potential on the molecular surface (where red indicates negative, blue indicates positive and green indicates neutral) |
When surface area and volume of ADV, PMEA, ADMP and ADDV are compared, it is found that the values for the pharmacologically active molecule ADDP are distinctly different from those of other compounds (Table 1) so that ADV, PMEA and ADMP may not be substrate for the key receptor.
CONCLUSION
Molecular modelling analyses based on semi-empirical and DFT calculations show that ADV and all its metabolites have large LUMO-HOMO energy differences so that they would be kinetically inert. This means that although all the compounds have some electron-deficient regions on the molecular surface so that they could react with glutathione and nucleobases in DNA, in actual fact the rate of such adverse reactions may not be significant because of the high kinetic inertness of the molecules.
ACKNOWLEDGMENTS
Fazlul Huq is grateful to the School of Biomedical Sciences, The University of Sydney for the time release from teaching.
REFERENCES
- Balzarini, J., Z. Hao, P. Herdewijn, D.G. Johns and E. De Clercq, 1991. Intracellular metabolism and mechanism of anti-retrovirus action of 9-(2-phosphonylmethoxyethyl)adenine, a potent anti-human immunodeficiency virus compound. Proc. Natl. Acad. Sci. USA., 88: 1499-1503.
Direct Link - Naesens, L., J. Balzarini, N. Bischofberger and E. De Clercq, 1996. Antiretroviral activity and pharmacokinetics in mice of oral bis(pivaloyloxymethyl)-9-(2-phosphonylmethoxyethyl)adenine, the bis(pivaloyloxymethyl) ester prodrug of 9-(2-phosphonylmethoxyethyl)adenine. Antimicrob. Agents Chemother., 39: 22-28.
- Srinivas, R.V., B.L. Robbins, M.C. Connelly, Y.F. Gong, N. Bischofberger and A. Fridland, 1993. Antiretroviral activity and pharmacokinetics in mice of oral bis(pivaloyloxymethyl)-9-(2-phosphonoylmethoxyethyl)adenine, the bis(pivaloyloxymethyl) ester prodrug of 9-(2-phosphonylmethoxyethyl)adenine. Antimicrob. Agents Chemother., 40: 22-28.