
Afshin Salavati
Department of Agronomy and Plant Breeding, Ramin Agriculture and Natural Resources University (Khouzestan), Iran
Alireza Taleei
Department of Agronomy and Plant Breeding, College of Agriculture and Natural Resources, University of Tehran, Karaj 31587-77871, Iran
Ali Akbar Shahnejat Bushehri
Department of Agronomy and Plant Breeding, College of Agriculture and Natural Resources, University of Tehran, Karaj 31587-77871, Iran
Alireza Abbasi
Department of Agronomy and Plant Breeding, College of Agriculture and Natural Resources, University of Tehran, Karaj 31587-77871, Iran
Payman Abbaszade Dahaji
Department of Soil Science, College of Agriculture and Natural Resources, University of Tehran, Karaj 31587-77871, Iran
Mahtab Omidvari
Department of Plant Protection, College of Agriculture and Natural Resources, University of Tehran, Karaj 31587-77871, Iran
Sohrab Rahimi
Department of Horticulture, College of Agriculture and Natural Resources, Tarbiat Modares University, Tehran, Iran
Journal of Plant Sciences
Year: 2011 | Volume: 6 | Issue: 4 | Page No.: 155-164







ABSTRACT
Legume-rhizobia symbiosis is a biological process which because of their crucial role in introduction of fixed nitrogen into both agricultural and natural systems has been extensively studied but the molecular mechanisms programmed in legumes such as perception of Nod factors, subsequent signal transduction and nodule organogenesis are still unknown, especially in common bean. In this study, responses of mitochondorial proteins of Phaseolus vulgaris L. root cells under early stages of interaction with Rhizobium etli was studied using shotgun proteomics technique in a time course manner. Three-day-old bean seedlings were infected by bacteria. At 0, 12, 24, 48 and 72 h after inoculation, the plant roots were collected, mitochondoria was purified and proteins were extracted and analyzed by mass spectrometry. Out of 15 proteins identified, up-regulated proteins were related to energy and disease and defence; whereas down-regulated proteins were related to protein synthesis and protein destination. These results provide an evidence to decrease in metabolism and signalling mechanism in root during early stages of symbiosis.
PDF Abstract XML References Citation
Received: August 05, 2011;
Accepted: November 12, 2011;
Published: December 21, 2011
How to cite this article
Afshin Salavati, Alireza Taleei, Ali Akbar Shahnejat Bushehri, Alireza Abbasi, Payman Abbaszade Dahaji, Mahtab Omidvari and Sohrab Rahimi, 2011. Comprehensive Expression Analysis of Time-dependent Responses of Mitochondrial Proteins in Phaseolus vulgaris L. Root Cells to Infection with Rhizobium etli. Journal of Plant Sciences, 6: 155-164.
DOI: 10.3923/jps.2011.155.164
URL: https://scialert.net/abstract/?doi=jps.2011.155.164
DOI: 10.3923/jps.2011.155.164
URL: https://scialert.net/abstract/?doi=jps.2011.155.164
INTRODUCTION
Legumes can establish intracellular interactions with symbiotic as well as pathogenic microbes. Such intracellular accommodation of bacteria always leads to the formation of a membrane compartment - the interface between the cytoplasm of the host and the bacteria (Ivanov et al., 2010). During early stages of symbiosis, the plant cells and the microsymbionts experience the specific structures and metabolic functions, which are essential for effective and stable nitrogen fixation (Samac and Graham, 2007). The rhizobia enter the deformed root hairs by endocytosis-like event and are surrounded by a new subcellular structure called infection thread that is derived from the plasma membrane of the root-hair cells (Brewin, 2004). It is believed that microaerobic conditions in the infected zone leads to morphological changes of mitochondria including the larged and elongated shape of mitochondria and intensive folding of the inner membranes (Werner and Morschel, 1978). These alterations might be related to metabolic modification of mitochondria to generate large amounts of organic acids such as C4-dicarboxylic acids, to conform the large demand for bacteroid respiration (Tajima and Kouchi, 1995). The study of isolated mitochondria from nodules showed higher oxidative and phosphorylative enzyme activities in comparison with this organell from other parts of roots (Suganuma and Yamamoto, 1987). Although, understanding of plant responses to compatible bacteri has increased, very little biochemical evidence has been reported on mitochondria differentiation in this symbiotic condition.
Recently, proteome analyses became a powerful tool for the investigation of complex cellular processes (Cheng et al., 2010) and were also successfully used for genetic and physiological studies in plants (Kaufmann et al., 2011). In plant biology, Proteome research often is based on a subset of proteins of eukaryotic cells called subproteome (Cordewell et al., 2000; Jung et al., 2000) and prosperous subproteomic analyses were carried out for the cell wall (Nilsson et al., 2010), the plasma membrane (Hynek et al., 2009), mitochondoria (Lister et al., 2004) and the thylakoids (Pribil et al., 2010) that led to investigate the hundreds proteins in which several of them were identified for the first time. Hoa et al. (2004) by using proteomic analysis on mitochondria isolated from soybean nodules reported high specific activity for proteins participated in electron transport and ATP synthesis, processing and translocating mitochondrial proteins, pyruvate decarboxylation and tricarboxylic acid cycle and mitochondria protection against oxidative stress in soybean nodule mitochondria. Although most proteomic studies are based on the separation of protein mixtures by two-dimensional gel electrophoresis (2-DE) and subsequent identification of the resolved proteins by sequencing or mass spectrometry, the resolution capacity of 2-DE is still insufficient to monitor entire protein sets of organelles (Rabilloud and Lelong, 2011).
In order to obtain a better insight into the molecular mechanism of early events of symbiosis in common bean with bacteria and also to determine which putative proteins from the common bean genome sequence represent constitutively expressed proteins for mitochondrial function, we have used shotgun proteomics analysis on purified common bean mitochondria. This approach aims to identify symbiosis related proteins localized in plant mitochondria, thus providing new insights into the function of plant mitochondria.
MATERIAL AND METHODS
Bacteria and plant conditions: Rhizobium etli biovariety Phaseoli was grown at 30°C for 3 days in yeast mannitol broth medium containing 3.67 mM K2HPO4, 0.81 mM MgSO4, 1.71 mM NaCl, 50 mM mannitol, 40 mM CaCl2 and 0.04% yeast extract adjusted to pH 6.8 (Hooykaas et al., 1977). Before plant inoculation, R. etli cells were centrifuged for 10 min with 2000 g and washed and diluted with sterile water to OD600 = 0.1.
Common bean (Phaseolus vulgaris L.) cultivar Akhtar was used. Seeds were sterilized in 2% sodium hypochlorite and rinsed several times with sterile distilled water and germinated on sand under white fluorescent light (600 μmol m-2 sec-1, 16 h light period) in a growth chamber (Sanyo, Tokyo, Japan) maintained at 25°C and 70% relative humidity. Three-day-old seedlings were inoculated with R. etli suspension and water until all seedlings appeared evenly wet as inoculation or mock inoculation, respectively. For proteomics analysis, roots were collected at 0, 12, 24, 48 and 72 h after inoculation and immediately frozen in liquid nitrogen. All experiments were biologically repeated 3 times.
Mitochondrial isolation and purification: Mitochondria were isolated and purified from roots as described by Millar et al. (2001). The procedure included two centrifugation steps for purification by Percoll step gradient consisting of 1:4:2 ratios of 40:23:18 Percoll and 28% Percoll gradients. The purity of mitochondria was evaluated by assay the activities of marker enzymes including cytochrome c oxidase, catalase, alkaline pyrophosphatase and alcohol dehydrogenase from each 2 mL fractions from top to bottom of the Percoll step gradients; according to Millar et al. (2001).
Protein extraction: Proteins were extracted from purified mitochondria fractions of roots by freeze-thawing in lysis solution containing 40 mM Tris-HCl (pH 7.5), 50 mM DTT and 2% (w/v) TritonX-100 and precipitated in 80% (v/v) acetone at -28°C for 2 h (Hoa et al., 2004).
Peptide preparation for mass spectrometry analysis: To prepare samples for shotgun proteomics, a ReadyPrep 2D cleanup kit (Bio-Rad) was used to further purify the mitochondorial protein pellet. The pellets were incubated in denaturing solution containing 8 M urea, 0.1 M NH4HCO3 and 0.1 M EDTA (pH 8.5), reducted in solution containing 30 mM DTT at 37°C for 90 min, alkylated in 50 mM iodoacetamide for 30 min and then digested with 1 mL of 10 pM trypsin at 37°C for 16 h followed by concentration and desalting with a NuTip C-18.
Protein identification by nano-liquid chromatography-tandem mass spectrometry: A nanospray LTQ XL Orbitrap MS (Thermo Fisher Science, San Jose, CA, USA) was operated in data-dependent acquisition mode with the installed XCalibur software. Peptides in 0.1% formic acid were loaded onto a 300 μm IDx5 mm C18 PepMap trap column by using an Ultimate 3000 nanoLC (Dionex, Germering, Germany). The peptides were eluted from the trap column and their separation and spraying were done using 0.1% formic acid in acetonitrile at a flow rate of 200 nL min-1 on a nano-capillary column (NTTC-360/75-3, Nikkyo Technos, Tokyo, Japan) with a spray voltage of 1.8 kV. Fullscan mass spectra were acquired in the Orbitrap over 150-2000 m/z with a resolution of 15000. The three most intense ions above the 1000 threshold were selected for collision-induced fragmentation in the linear ion trap at normalized collision energy of 35% after accumulation to a target value of 1000. Dynamic exclusion was employed within 30 s to prevent repetitive selection of peptides.
Differential expression analysis was performed using SIEVE software (ver. 1.1.0, Thermo fisher) on raw spectral data files generated by MS. False discovery rate for peptide matches above identity threshold for the datasets was less than 5% according to the mascot search. To validate the p-value in SIEVE, duplicates of three individual biological samples were used. For frame creation, m/z ranges were set to 500-1500 and retention time to 10-115 min. Frame m/z width was 0.02 amu and frame time width was 2.5 min. The threshold parameter was set to 500000. Using SEQUEST search engine in SIEVE software, soybean database searching was performed with a maximum of 10000 frames to be searched and proteins with significant changes in peak intensities were identified, based on the results of comparative analysis.
Functional assignment of proteins: Identified proteins were annotated to their biological function according to Bevan et al. (1998). Information obtained from Universal Protein Resource (http://www.uniprot.org/), Phytozome (http://www.phytozome.net/) was used for classification.
Clustering and statistical analyses: For clustering analysis, the log2-transformed ratios of peak intensities of protein were used. Hierarchical clustering of proteins based on their intensities profiles was performed using Gene Cluster 3.0 software (De Hoon et al., 2004) with Euclidean distance similarity metric and complete linkage method. The resulting clusters were visualized using JAVA TREEVIEW software (Saldanha, 2004). The statistical significance of the results was evaluated with the Student's t-test when only two means were compared or with two-way ANOVA followed by Duncan's multiple comparisons test otherwise. Data analyses and graphical representations were performed using Microsoft Office Excel 2007 or R, a language and environment for statistical computing and graphics (Ihaka and Gentleman, 1996).
RESULTS
Isolation of mitochondria from common bean root cells in high-purity level: The reliability of a mitochondria proteome, also other subcellular proteomes, is largely dependent on the purity of the isolated organelle away from other cellular contaminants. In this study, two Percoll gradient density separations were used that leads to gain the mitochondria free of contamination by cytosol, peroxisomes, plastids and other organelles (Fig. 1).
On the first step-gradient, mitochondria band was located at the interface of the 23 and 40% Percoll steps (in fractions 11-14; Fig. 1a). Altough, the activity of catalase and alkaline pyrophosphatase this region implying the contamination by peroxisomes and plastids, respectively. However, the lack of alcohol dehydrogenase in these fractions indicates the absence of contamination by cytosol. To more purification of mitochondria, a second gradient containing of 28% Percoll was used (Fig. 1b). Corresponding to activity of cytochrome c oxidase at fraction 3, a broad opaque band in the upper part of the gradient is formed by mitochondria.
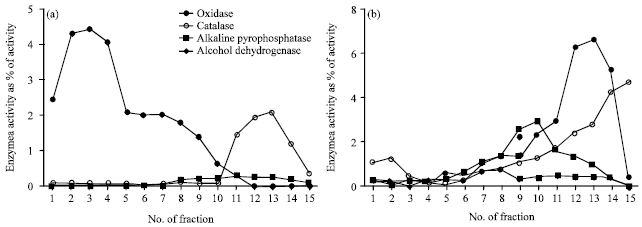 | |
| Fig. 1(a-b): | Isolation of common bean mitochondria by density centrifugation. The organelle pellet from homogenized common bean root cells was loaded onto a Percoll step gradient consisting 18:23: 40% Percoll correspondence to fractions 1-4: 5-12: 13-15, respectively (a). For second density centrifugation, mitochondria were recovered from the 40:23% interface (fractions 11-14) and were loaded onto a Percoll gradient containing 28% Percoll (b). Two-milliliter fractions were collected from top to bottom of both gradients and the activities of cytochrome c oxidase, catalase, alkaline pyrophosphatase and alcohol dehydrogenase were assayed in each fraction. Values are expressed as a percentage of the total activity in the initial cell extract |
Altough catalase activity was formed a peak lower down the gradient between fractions 11-14 but catalase activity was not concomitant with cytochrome c oxidase activity, this result confirm that this band was free of contamination by peroxisomes.
Fifteen proteins are changed under early stages of symbiosis as identified by shotgun proteomics technique: Tryptic digested peptides were eluted and separated in a nano-LC trap column and sprayed in MS. Individual searches of converted data files against NCBI database resulted about 390 protein hits with negligible variations among samples. In this study, SIEVE software was applied to demonstrate automated analysis of differential proteomic expression profiling. Three biological replicates were applied twice in MS and generated raw data files were used for label-free differential analysis. All chromatograms were aligned to the one of the controls as reference control and total of 40858 peaks were detected. According to the SEQUEST result, 15 significantly changed proteins were identified, of which 9 proteins were upregulated and 6 proteins were downregulated. Using SIEVE software the ratio of up- and downregulated proteins were calculated in which the values >1 indicates upregulated proteins (Table 1). Upregulated proteins were aconitate hydratase, isocitrat dehydrognase, citrate synthase precursor, nucleoside diphosphate kinase, peroxyredoxin, glutathion peroxidase, malat dehydrogenase, resistance like protein and Gly-rich RNA-binding PsGRBP whereas down-regulated proteins were aminoacyl-tRNA synthetase, chaperonin, porin/voltage dependent anion-selectiv channel protein, translation elongation factor and succinyl-coenzyme A ligase. Classification of the identified proteins based on the protein function revealed that must up-regulated proteins were categorized as energy and disease/defense related proteins. Interestingly, the proteins which classified in protein destination and protein synthesis were downregulated under early stages of symbiosis (Fig. 2).
| Table 1: | Mitochondorial proteins with significant changes in expression level under early stages of symbiosis identified by LC MS/MS-based proteomics using SIEVE (p<0.05) |
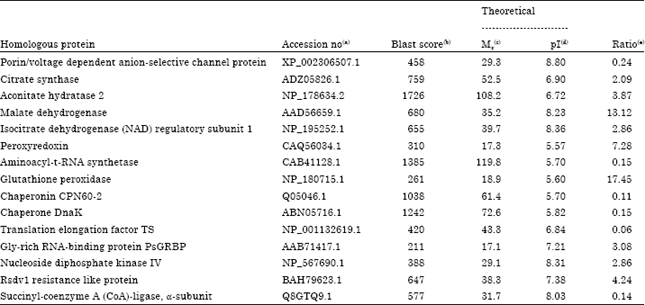 | |
| (a)Accession no., accession number according to the BLAST search in NCBI database. (b)Blast score, score of the BLAST search in NCBI database. (c)Mr, molecular weight. (d)pI, isoelectric point. (e)Ratio, protein abundance changes at 72 h after inoculation in compared to the control. Proteins with the ratio greater than 1 are up-regulated | |
Analysis of expression profiles of differentially expressed protein during early stages of symbiosis: To find trends in the identified differentially expressed proteins, hierarchical clustering analysis was performed with expression data of the 15 proteins in 0, 12, 24, 48 and 72 h treatments. The clustering divided the 15 protein into 6 clusters (Fig. 3). Proteins belonged to cluster 1 were significantly increased in all treatments in comparison with control at same level, indicating that these proteins are increased by experience by start the symbiosis. Proteins belonged to cluster 2 were increased in each treatments more than before step, indicating that increase of these proteins were associated with duration of symbiosis.
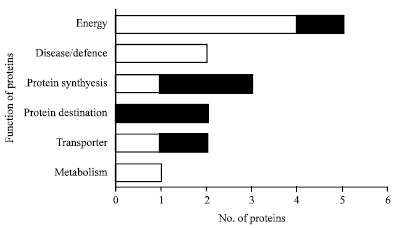 | |
| Fig. 2: | Classification of the differentially changed proteins during early stages of symbiosis. After identification of differentially expressed proteins during early stages of symbiosis, functions of identified proteins were assigned using classification described by Bevan et al. (1998). Proteins were classified in 6 groups |
 | |
| Fig. 3: | Expression profile of identified proteins during early stages of symbiosis. Identified proteins were categorized in 6 classes based on change in protein expression level during early stages of symbiosis. Classes 1-3 and 4-6 represented up- and down-regulated proteins, respectively. In up-regulation category, continuously upward expression and significantly up-regulation at 1DFE in comparison with control, were mentioned in classes 1, 2 and 3, respectively. In down-regulation category, continuously downward expressions and significantly down-regulation at 1DFE in comparison with control were mentioned in classes 4, 5 and 6, respectively |
Protein belonged to cluster 3 were increased in 12 and 72 h after inoculation more than internal treatment, indicating that this proteins do not change depends on the duration of symbiosis. Proteins belonged to cluster 4 were gradually decreased from 12 h to 72 h, indicating that these proteins are decreased depending on duration of symbiosis. Proteins belonged to cluster 5 were at same levels in all treatments, indicating that expression of these proteins disturbed by symbiosis and expression of them is not depends on the duration of symbiosis. Protein belonged to cluster 6 was decreased in each treatments more than before step, indicating that decrease of these proteins were associated with duration of symbiosis.
DISCUSSION
A number of investigations have presented the mitochondrial proteome study from soybean (Hoa et al., 2004), rice (Millar et al., 2004), pea (Taylor et al., 2005) and Arabidopsis (Heazlewood et al., 2003). Hoa et al. (2004) identified 17 mitochondrial associated proteins in soybean mitochondorial preparations using a combination of N-terminal sequencing and MS/Ms based sequencing. Most of these studies have reported changes in proteome of different tissues from the same plant under same or different stresses and/or during developmental changes. However, identifications of the different protein have been limited by the need for cross-reacting antibodies or sufficient material and finance for N-terminal sequencing. Present study show that using the available full genome sequence of a related family of non-model plants can enables the high-throughput identifications of plant organell proteins from MS data by inexpensively procedure. We have used a extracted mitochondoria from roots under interaction with bacteria in a time course mannner in our experiments.
Purity of mitochondria proteins: Regarding to, plastids classification into three separate groups (Pyke, 2010), carotenoids, as a regularly marker for plastids, is correlated with only one of the plastid peaks (Millar et al., 2001). Thus, the existence of carotenoids is not a reliable marker for the presence of all types of plastids. A slight contamination by plastids were expected acoording to a trifle peak of alkaline pyrophosphatase activity in fractions 2-4. Fortunately, this level of contamination was sufficiently minor therfore no differentially changed proteins under symbiosis with plastid origin were detected in this study. Any proteins involved in photosystem, the Calvin cycle, or major peroxisomal proteins (Table 1) have not identified.
Identifying functions in common bean mitochondria under symbiosis: In order to identify the mechanism involved in early stage of symbiosis between common bean and bacteria, mitochondorial proteins of common bean roots were analyzed at 0, 12, 24, 48 and 72 h after inoculation. Out of down-regulated proteins, the major functional category was protein destination/storage which are chaperone DnaK and cpn 60 chaperonin. Yeh et al. (2002) reported that change in expression of chaperonins occure under early stage of symbiosis. Chaperonins are a ubiquitous family of abundant proteins which presumably participate in protein synthesis, folding, degradation (Boston et al., 1996) and transporting proteins across membranes and the formation of organelles (Vierling, 1991). In this study, chaperonin families including chaperone DnaK and cpn60 chaperonin were down-regulated be symbiosis which suppression of chaperone DnaK was occure from beginning of interaction whereas suppression of cpn60 were occure only in advanced satges (Fig. 3). The change of chaperone level in symbiosis (Oehrle et al., 2008) as well as response to pathogen (Colditz et al., 2004) was reported and points out a function in repair and degradation processes during responses of plant cell to biotic stress. chaperone have also been identified in the symbiosome membrane of soybean (Panter et al., 2000), Lotus japonicas (Wienkoop and Saalbach, 2003) and Medicago truncatula (Catalano et al., 2004). Protein degradation can be part of the normal cellular protein turnover process but can also play an important role in the control of plant development and plant-microbe interactions (Hellmann and Estelle, 2002). Change in chaperonin levels under symbiosis interaction was reported in Medicago truncatula (Lei et al., 2005) and soybean. Given the possible role of chaperonins in pathogen-host interactions, this results suggest that down-regulated chaperonins might leads to decrease in protein degradation.
Protein related to energy was a big category of identified proteins which included isocitrate dehydrogenase, malate dehydrogenase, citrate synthase and aconitate hydratase 2 were up-regulated under early stages of symbiosis. The induction ratio of isocitrate dehydrogenase and citrate synthase was same and they moderately induction were recorded for them under all steps of experiment whereas malate dehydrogenase showed a high level of expression fro beginning of interaction (Fig. 2). The enzymes of the TCA cycle including isocitrate dehydrogenase, malate dehydrogenase and citrate synthase were up-regulated in common bean roots under early stages of symbiosis. Isocitrate dehydrogenase is an enzyme of the TCA cycle. Although annotation of isocitrate dehydrogenase as an NADP-dependent enzyme suggests a mitochondrial localization, this enzyme was not identified proteins of soybeans mitochondria (Hoa et al., 2004). However, induction of isocitrate dehydrogenase has been identified in the root of soybean roots (Oehrle et al., 2008) and bacteroids (Sarma and Emerich, 2005). Kurz and Larue (1977) reported that isocitrate dehydrogenase activity in bacteroids was found to be highest when nitrogen fixation was at a maximum. Malate dehydrogenase is another identified TCA related enzyme that was up-regulated in this study. Up regulation of malate dehydrogenase under symbiosis were reported Medicago truncatula (El-Yahyaoui et al., 2004), soybean (Hoa et al., 2004; Oehrle et al., 2008) and Lotus japonicas (Wienkoop and Saalbach, 2003). These results suggest that the increase in concentration of TCA cycle intermediates probably is needed to increase the flux of the TCA cycle and meet the increased energy demand of establish the new organ in roots.
Differentially expressed proteins classified into disease/defence category with exception of ripening related protein were up-regulated under early stages of symbiosis. The proteins in disease/defence category were peroxyredoxin and glutathione peroxidase. peroxiredoxin has antioxidant activity and might function in the removal of hydrogen peroxide. It is demonstrated that symbiotic interaction requires temporal and spatial activation of different defence mechanisms in infected plants (Garcia-Garrido and Ocampo, 2002), therefore, changes in protein levels involved in disease/defence mechanisms could partly explain that early stage of symbiosis between common bean and rhizobia could resemble a pathogen attack and mechanisms involved to response to stress might be induced in early stage of symbiosis.
ACKNOWLEDGMENTS
The authors are grateful to scholarship section of the Ministry of Science, Research and Technology of Islamic Republic of Iran and the Higher Education Department of University of Tehran.
REFERENCES
- Bevan, M., I. Bancroft, E. Bent, K. Love and H. Goodman et al., 1998. Analysis of 1.9 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature, 391: 485-488.
CrossRefDirect Link - Brewin, N.J., 2004. Plant cell wall remodelling in the rhizobium-legume symbiosis. Crit. Rev. Plant Sci., 23: 293-316.
CrossRefDirect Link - Catalano, C.M., W.S. Lane and D.J. Sherrier, 2004. Biochemical characterization of symbiosome membrane proteins from Medicago truncatula root nodules. Electrophoresis, 25: 519-531.
CrossRefPubMedDirect Link - Cheng, Z., B.J. McConkey and B.R. Glick, 2010. Proteomic studies of plantebacterial interactions. Soil Biol. Biochem., 42: 1673-1684.
CrossRef - Colditz, F., O. Nyamsuren, K. Niehaus, H. Eubel, H.P. Braun and F. Krajinski, 2004. Proteomic approach: Identification of Medicago truncatula proteins induced in roots after infection with the pathogenic oomycete Aphanomyces euteiches. Plant Mol. Biol., 55: 109-120.
CrossRefDirect Link - Cordewell, S.J., A.S. Nouwens, N.M. Verrills, D.J. Basseal and B.J. Walsh 2000. Subproteomics based upon protein cellular location and relative solubilities in conjunction with composite two-dimensional electrophoresis gels. Electrophoresis, 21: 1094-1103.
CrossRefPubMedDirect Link - De Hoon, M.J.L., S. Imoto, J. Nolan and S. Miyano, 2004. Open source clustering software. Bioinformatics, 20: 1453-1454.
CrossRef - El-Yahyaoui, F., H. Kuster, B. Ben Amor, N. Hohnjec and A. Puler et al., 2004. Expression profiling in Medicago truncatula identifies more than 750 genes differentially expressed during nodulation, including many potential regulators of the symbiotic program. Plant Physiol., 136: 3159-3176.
CrossRefPubMedDirect Link - Garcia-Garrido, J.M. and J.A. Ocampo, 2002. Regulation of the plant defence response in arbuscular mycorrhizal symbiosis. J. Exp. Bot., 53: 1377-1386.
CrossRefDirect Link - Heazlewood, J.L., K.A. Howell, J. Whelan and A.H. Millar, 2003. Towards an analysis of the rice mitochondrial proteome. Plant Physiol., 132: 230-242.
CrossRefDirect Link - Hellmann, H. and M. Estelle, 2002. Plant development: Regulation by protein degradation. Science, 297: 793-797.
CrossRefDirect Link - Hoa, L.T.P., M. Nomura and S. Tajima, 2004. Characterization of bacteroid proteins in soybean nodules formed with Bradyrhizobium japonicum USDA110. Microb. Environ., 19: 71-75.
CrossRefDirect Link - Hooykaas, P.J.J., P.M. Klapwijk and M.P. Nuti, 1977. Transfer of the Agrobacterium tumefaciens TI plasmid to avirulent agrobacteria and to rhizobium ex planta. Microbiology, 98: 477-484.
CrossRefDirect Link - Hynek, R., B. Svensson, O.N. Jensen, V. Barkholt and C. Finnie, 2009. The plasma membrane proteome of germinating barley embryos. Proteomics, 9: 3787-3794.
CrossRefPubMedDirect Link - Ihaka, R. and R. Gentleman, 1996. R: A language for data analysis and graphics. J. Comput. Graphical Stat., 5: 299-314.
CrossRefDirect Link - Ivanov, S., E. Fedorova and T. Bisseling, 2010. Intracellular plant microbe associations: Secretory pathways and the formation of perimicrobial compartments. Curr. Opin. Plant Biol., 13: 372-377.
PubMed - Jung, E., M. Heller, J.C. Sanchez and D.F. Hochstrasser, 2000. Proteomics meets cell biology: The establishment of subcellular proteomes. Electrophoresis, 21: 3369-3377.
PubMed - Kaufmann, K., C. Smaczniak, S. De-Vries, G.C. Angenent and R. Karlova, 2011. Proteomics insights into plant signaling and development. Proteomics, 11: 744-755.
CrossRef - Kurz, W.G. and T.A. Larue, 1977. Citric acid cycle enzymes and nitrogenase in nodules of Pisum sativum. Can. J. Microbiol., 23: 1197-1200.
PubMed - Lister, R., O. Chew, M.N. Lee, J.L. Heazlewood and R. Clifton et al., 2004. A transcriptomic and proteomic characterization of the Arabidopsis mitochondrial protein import apparatus and its response to mitochondrial dysfunction. Plant Physiol., 134: 777-789.
CrossRef - Millar, A.H., A.E. Trend and J.L. Heazlewood, 2004. Changes in the mitochondrial proteome during the anoxia to air transition in rice focus around cytochrome-containing respiratory complexes. J. Biol. Chem., 279: 39471-39478.
CrossRef - Millar, A.H., L.J. Sweetlove, P. Giege and C.J. Leaver, 2001. Analysis of the Arabidopsis mitochondria proteome. Plant Physiol., 127: 1711-1727.
PubMed - Nilsson, R., K. Bernfur, N. Gustavsson, G. Wingsle and C. Larsson, 2010. Proteomics of plasma membranes from poplar trees reveals tissue distribution of transporters, receptors, and proteins in cell wall formation. Mol. Cell. Proteomics, 9: 368-387.
CrossRef - Oehrle, N.W., A.D. Sarma, J.K. Waters and D.W. Emerich, 2008. Proteomic analysis of soybean nodule cytosol. Phytochemistry, 69: 2426-2438.
PubMed - Panter, S., R. Thomson, G. De-Bruxelles, D. Laver, B. Trevaskis and M. Udvardi, 2000. Identification with proteomics of novel proteins associated with the peribacteroid membrane of soybean root nodules. Mol. Plant Microbol. Interact., 13: 325-333.
CrossRef - Rabilloud, T. and C. Lelong, 2011. Two-dimensional gel electrophoresis in proteomics: A tutorial. J. Proteomics, 74: 1829-1841.
CrossRefPubMedDirect Link - Saldanha, A.J., 2004. Java treeview-extensible visualization of microarray data. Bioinformatics, 20: 3246-3248.
CrossRefPubMedDirect Link - Samac, D.A. and M.A. Graham, 2007. Recent advances in legume-microbe interactions: Recognition, defense response and symbiosis from a genomic perspective. Plant Physiol., 144: 582-587.
CrossRef - Sarma, A.D. and D.W. Emerich, 2005. Global protein expression pattern of Bradyrhizobium japonicum bacteroids: A prelude to functional proteomics. Proteomics, 5: 4170-4184.
PubMed - Taylor, N.L., J.L. Heazlewood, D.A. Day and A.H. Millar, 2005. Differential impact of environmental stresses on the pea mitochondrial proteome. Mol. Cel. Proteomics, 4: 1122-1123.
CrossRef - Vierling, E., 1991. The roles of heat shock proteins in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol., 42: 579-620.
CrossRefDirect Link - Werner, D. and E. Morschel, 1978. Differentiation of nodules of Glycine max. Planta, 141: 169-177.
CrossRef - Wienkoop, S. and G. Saalbach, 2003. Proteome analysis. Novel proteins identified at the peribacteroid membrane from Lotus japonicus root nodules. Plant Physiol., 131: 1080-1090.
CrossRef - Yeh, K.C., M.C. Peck and S.R. Long, 2002. Luteolin and GroESL modulate in vitro activity of NodD. J. Bacteriol., 184: 525-530.
PubMedDirect Link - Suganuma, N. and Y. Yamamoto, 1987. Respiratory metabolism of mitochondria in soybean root nodules. Soil Sci. Plant Nutr., 33: 93-101.
Direct Link - Pribil, M., P. Pesaresi, A. Hertle, R. Barbato and D. Leister, 2010. Role of plastid protein phosphatase tap38 in lhcii dephosphorylation and thylakoid electron flow. PLoS Biol., Vol. 8.
CrossRef