
Xiao Yao
Department of Chemistry, Fudan University, Shanghai 200433, China
Jianxia Zhang
Department of Chemistry, Fudan University, Shanghai 200433, China
Gang Chen
Department of Chemistry, Fudan University, Shanghai 200433, China
Journal of Plant Sciences
Year: 2007 | Volume: 2 | Issue: 3 | Page No.: 273-282







ABSTRACT
Capillary electrophoresis with electrochemical detection has been employed for the determination of three phenolic acetophenones (including paeonol, p-hydroxyacetophenone and 2,4-hydroxyacetophenone) in Radix Cynanchi Paniculati (i.e., Paniculate Swallowworf Root) for the first time. Effects of several important factors such as the concentration and the acidity of the running buffer, separation voltage, injection time and detection potential were investigated to acquire the optimum conditions. The detection electrode was a 300 μm diameter carbon disc electrode at a working potential of +0.90 V (vs. Saturated Calomel Electrode (SCE)). The three analytes can be well separated within 10 min in a 50 cm length fused silica capillary at a separation voltage of 15 kV in a 50 mM borate buffer (pH 9.2). The relation between peak current and analyte concentration was linear over about 3 orders of magnitude with the detection limits (S/N = 3) ranging from 0.20 to 0.36 μM for all analytes. The proposed method has been successfully applied to the determination of the bioactive constituents in the real herbal samples with satisfactory assay results.
PDF Abstract XML References Citation
How to cite this article
Xiao Yao, Jianxia Zhang and Gang Chen, 2007. Determination of Phenolic Acetophenones in Radix Cynanchi Paniculati by Capillary Electrophoresis with Electrochemical Detection. Journal of Plant Sciences, 2: 273-282.
DOI: 10.3923/jps.2007.273.282
URL: https://scialert.net/abstract/?doi=jps.2007.273.282
DOI: 10.3923/jps.2007.273.282
URL: https://scialert.net/abstract/?doi=jps.2007.273.282
INTRODUCTION
As an important traditional Chinese medicine, Radix Cynanchi Paniculati (i.e., Paniculate Swallowworf Root) is the dried root or rhizome of Cynanchum paniculatum (Bunge) Kitag., which belongs to the Asclepiaaaceae family. It can expel wind, promote blood circulation and remove blood stasis, alleviate pain, relieve cough and dyspnea and clear away toxins and relieve swe1ling (Pharmacopoeia of People’s Republic of China, 2005). Radix Cynanchi Paniculati has been frequently used as an important ingredient in many traditional prescriptions (Pharmacopoeia of People’s Republic of China, 2005; Jiang, 1994). It is also an important material for the extraction of paeonol in the medicinal industry. Paeonol is the main effective constituent of Radix Cynanchi Paniculati. It has been proved by modern pharmacology that paeonol has a broad range of physiological activities, such as anti-bacteria, anti-inflammation, anti-allergy, alleviating pain and enhancing the immune system (Jiang and Chen, 1994; Chou, 2003; Kim et al., 2004; Chinese Materia Medica, 1999). Paeonol has found wide applications in medicine, incense and chemistry. It has been used clinically as a therapeutic medicine for myalgia, rheumatic pain, neuralgia, coetaneous pluribus, etc. (Jiang and Chen, 1994). The paeonol content is an important parameter for evaluating the quality of Radix Cynanchi Paniculati. The Pharmacopoeia of China requires the content of paeonol in Radix Cynanchi Paniculati to be no less than 1.3% (Pharmacopoeia of People’s Republic of China, 2005). Hence, it is interesting to establish some rapid, simple and accurate approaches for the determination of paeonol and some coexistent constituents in Radix Cynanchi Paniculati.
Several methods have been developed to determine paeonol and some coexistent substances in Radix Cynanchi Paniculati, such as Liquid Chromatography (LC) (Pharmacopoeia of People’s Republic of China, 2005; Zhan and Li, 2004; Yu, 2005), Gas Chromatography (GC) (Deng et al., 2006; Guo et al., 1996), Thin Layer Chromatography (TLC) (Zhang, 1989), etc. Separation and determination of various constituents in plant drugs is always a complicated and challenging task. Since Capillary Electrophoresis (CE) in its modern form was first described by Jorgenson and Luckas (1981a, b) the application of Capillary Electrophoresis (CE) for the separation of various bioactive constituents in medicinal plants has become increasingly widespread because of its minimal sample volume requirement, short analysis time and high separation efficiency (Peng et al., 2005; Chu et al., 2004). CE analysis holds considerable promise for biomedical and pharmaceutical analysis, clinical diagnostics, environmental monitoring and forensic investigations (Piergiovanni and Taranto, 2005; Lin et al., 2003; Huck et al., 2005; Guan et al., 2006; Fliser et al., 2005). Electrochemical Detection (ECD) typically operated in the amperometric mode can be coupled with CE to provide high sensitivity and selectivity for the determination of electroactive substances in herbal drugs (Holland and Leigh, 2002; Baldwin, 2000). It offers great promise for CE, with features that include high sensitivity, inherent miniaturization of both the detector and control instrumentation, low cost, low-power demands and high compatibility with micromachining technologies (Holland and Leigh, 2002). Nowadays, it is of high importance to control the quality of herbal drugs based on their bioactive constituents and some co-existent substances. In 2000, the US Food and Drug Administration (FDA) published a draft of Guidance for Industry Botanical Drug Products. Before a plant drug can become legally marketed, its spectroscopic or chromatographic fingerprints and chemical assay of the characteristic markers are required. CE-ECD should find more applications in this area.
CE-ECD should be an assistant, alternative and complement technique for the determination of paeonol (PN), p-hydroxyacetophenone (PHA) and 2,4-hydroxyacetophenone (DHA) in Radix Cynanchi Paniculati because the three phenolic acetophenones all contain phenolic hydroxyl groups that are electroactive at modest oxidation potential on carbon electrode. Moreover, ECD can provide higher selectivity as only electroactive substances can be detected so that the electropherograms are greatly simplified, which is important for the analysis of medicinal plants because their constituents are usually complex. In this study, CE-ECD was employed for the determination of PN, PHA and DHA (their molecular structures are shown in Fig. 1) in Radix Cynanchi Paniculati without derivatization.
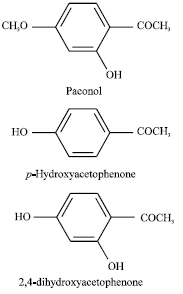 | |
| Fig. 1: | Molecular structure of paeonol (PN), p-hydroxyacetophenone (PHA) and 2,4-hydroxyacetophenone (DHA) |
This method is simple, sensitive, selective and efficient, providing not only a way for evaluating the quality of Radix Cynanchi Paniculati in marketplaces, but also an alternative approach for quality control in medicinal factories and constituent investigations of other related plants. To our best knowledge, there are no earlier reports published on the determination of the bioactive constituents in Radix Cynanchi Paniculati by CE. The optimization, detailed characterization and advantages of the CE-ECD approach are reported in the following sections in connection to the measurement of phenolic acetophenones in the herbal drugs.
MATERIALS AND METHODS
Reagent and Solutions
Paeonol (PN), p-hydroxyacetophenone (PHA) and 2,4-hydroxyacetophenone (DHA) were all supplied by National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). All aqueous solutions were made up in doubly distilled water. Other chemicals were of analytical grade.
Stock solutions of PN, PHA and DHA (10 mM) were all prepared in a 50% (V/V) ethanol-water mixture and were kept in a 4°C refrigerator. They were stable for at least 1 month. The running buffer was 50 mM borate buffer (pH 9.2) unless mentioned otherwise. The stock solutions were diluted to the desired concentration with the running buffer just prior to use.
Apparatus
The CE-ECD system used has been described previously (Chen, 2001, 2002a, b and 2005). A±30 kV high-voltage dc power supply (Shanghai Institute of Nuclear Research, China) provided a separation voltage between the two ends of the capillary. The inlet of the capillary was held at a positive potential and the outlet of capillary was maintained at ground. The separations were carried out in a 50 cm length of 25 μm i.d. and 360 μm o.d. fused silica capillary (Polymicro Technologies, Phoenix, AZ, USA).
A three-electrode electrochemical cell consisting of a laboratory-made 300 μm diameter carbon disc working electrode, a platinum auxiliary electrode and a saturated calomel electrode (SCE) as the reference electrode, was used in combination with a BAS LC-4C amperometric detector (Bioanalytical Systems Inc., West Lafayette, IN, USA). The filter of the detector was set at 0.1 Hz. The working electrode was positioned carefully opposite the outlet of the capillary with the aid of a micromanipulator (CORRECT, Tokyo, Japan) and arranged in a wall-jet configuration. The distance between the tip of the working electrode and the capillary outlet was adjusted to ~50 μm by comparison with the bore (25 μm) in the capillary while being viewed under a microscope. The electropherograms were recorded using a LKB”REC 1 chart record (Pharmacia, Sweden). A YS 38-1000 220 V alternate constant-voltage power supply (Shanghai Instrumental Transformer Factory, Shanghai, China) was employed to suppress the voltage fluctuation of the power line. The whole system was assembled in a laboratory that was air-conditioned at 25°C to minimize the temperature fluctuation.
Sample Preparation
Three samples of Radix Cynanchi Paniculati were obtained from Sun-Tian-Tang Traditional Chinese Medicine Store (Shanghai, China). They were all dried at 60°C for 2 h and then were pulverized. About 1.0 g of the powder was weighed accurately and dispersed in 50 mL of methanol. The mixture was kept in a 60°C water bath for 3 h. After cooling, it was sonicated for 30 min and filtered through a filter paper. The extract was diluted using 50 mM borate buffer (pH 9.2) at a ratio of 10 (1 to 10) just prior to CE analysis.
Procedures
Before use, the carbon disc electrode was successively polished with emery paper and alumina powder and sonicated in doubly distilled water. The capillary used for the separation were treated before use by flushing with 0.1 M NaOH and doubly distilled water for 10 min each. Subsequently, the capillary was filled with the running buffer and was conditioned with the running buffer for at least 10 min at the voltage of 15 kV between the two ends of the capillary. CE was performed at a separation voltage of 15 kV, unless otherwise indicated. The potential applied to the working electrode was +0.90 V (vs. SCE). Samples were injected electrokinetically into the capillary at 15 kV for 6 sec. Before injection, both the anode end of the capillary and the platinum-wire anode were moved from the anode solution to the sample solution. After an injection voltage of 15 kV was applied between the two ends of the capillary for 6 sec, the sample solution could be introduced into the capillary. The anode end of the capillary together with the anode was then quickly returned to the anode solution. A voltage of 15 kV was subsequently applied in the constant-voltage mode for CE separation. The amperometric detector was on during the injection procedures. Note that the cathode solution in the electrochemical detection cell, the anode solution and the sample solution were all at the same level. Moreover, sample solutions, standard solutions and running buffer were all filtered through a polypropylene filter (0.22 μm, Shanghai Bandao Industry Co., Ltd., Shanghai, China) prior to their use. Peak identification was performed by the standard-addition method.
RESULTS AND DISCUSSION
Hydrodynamic Voltammograms (HDVs)
Paeonol, p-hydroxyacetophenone and 2,4-hydroxyacetophenone all contain phenolic hydroxyl groups, which are electroactive on carbon electrode. In this study, a carbon disc electrode was used for their detection. The potential applied to the working electrode directly affects the sensitivity and the detection limits of this method and it is necessary to determine the hydrodynamic voltammograms (HDVs) for the analytes to obtain the optimum potential. Figure 2 depicts the HDVs for the oxidation of the three phenolic acetophenone using the carbon disc-electrode detector. The curves were recorded pointwise from +0.1 to +1.1 V (vs. SCE) in steps of 0.1 V using a separation voltage of 15 kV. All analytes displayed similar profiles, with an increase of the response starting at +0.5 V (vs. SCE). The current rose rapidly upon raising the potential above +0.6 V (vs. SCE).
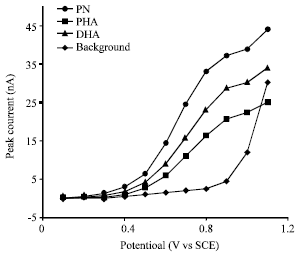 | |
| Fig. 2: | Hydrodynamic voltammograms (HDVs) for 0.25 mM of paeonol (PN), p-hydroxyacetophenone (PHA) and 2,4-hydroxyacetophenone (DHA) in CE. Fused-silica capillary: 25 μm i.d. χ 50 cm length; working electrode: 300 μm diameter carbon disc electrode; running buffer: 50 mM borate buffer (pH 9.2); separation and injection voltage: 15 kV; injection time: 6 sec |
Although an applied potential greater than +0.90 V (vs. SCE) resulted in higher peak currents, both the baseline noise and the background current increase substantially. The high background current led to an unstable baseline, which is a disadvantage for the sensitive and stable detection. Considering the sensitivity and background current, subsequent amperometric detection work employed a detection potential of +0.90 V (vs. SCE), which offered the most favorable signal-to-noise characteristics. The stability of the working electrode was good and the reproducibility was high at the optimum potential.
Effects of the Concentration and the pH of Buffer
In order to enhance the resolution and solubility of analytes, alkaline borate buffer was employed in this study. PN, PHA and DHA are partially negatively charged in alkaline borate buffer because their phenolic hydroxy groups can dissociate to form anions.
Because the buffer concentration influences the viscosity coefficient of the solution, the diffusion coefficient of analytes and the zeta-potential (ζ) of the inner surface of capillary tube as well, it affects not only the resolution and migration time of the analytes, but also the peak current. Figure 3A indicates that the migration time and the resolution increases with increasing buffer concentration when the pH was kept at 9.2. Upon raising the concentration above 50 mM, the peak current was low and the peak shape became poor because the electric current in the capillary also increased, resulting in Joule heating and peak broadening. In this experiment, a 50 mM borate buffer (pH 9.2) was chosen as the running buffer in considering the peak current, resolution, analytical time and the buffer capacity.
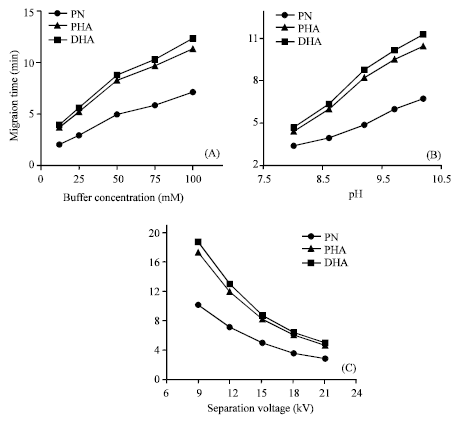 | |
| Fig. 3: | Effect of (A) the concentration and (B) the acidity of the running buffer and of (C) the separation voltage on the migration time of paeonol (PN), p-hydroxyacetophenone (PHA) and 2,4-hydroxyacetophenone (DHA). Working potential: +0.90 V (vs. SCE); other conditions were as in Fig. 2 |
The acidity of the running buffer affect the zeta-potential (ζ), the Electroosmotic Flow (EOF) as well as the overall charge of PN, PHA and DHA, which determine the migration time, peak height and the separation of the analytes (Chen, 2002). The effect of the running buffer pH on the migration time of PN, PHA and DHA is shown in Fig. 3B. The running buffers were 50 mM borate buffers at five different pH values (8.0, 8.6, 9.2, 9.7 and 10.2). As shown in Fig. 3B, the resolution of the three analytes is poor at pH 8.0. When the running buffer pH increases, the migration time increases with the resolution improved due to the dissociation of the hydroxyl groups for all analytes. Meanwhile, the peak current is low and the peak shape becomes poor at pH value above 9.7. At pH 9.2, PN, PHA and DHA can be well separated within a relatively short time. In this experiment, 50 mM borate buffer at pH of 9.2 was chosen as the running buffer in considering the peak current, resolution, the analytical time and the stability of the running buffer.
Effect of Separation Voltage and Injection Time
Figure 3C depicts the effect of separation voltage on the migration time for PN, PHA and DHA. Increasing the voltage gives shorter migration time for all the three analytes, but also increases the baseline noise, resulting in poorer detection limits. It is found that higher separation voltages are not beneficial to the resolution of PN, PHA and DHA and can result in higher Joule heating, which directly affects the separation efficiency of this method. Separation voltages, which are too low, will increase the analysis time considerably; this in turn causes peak broadening. On the basis of experiments, 15 kV was chosen as the optimum voltage to accomplish a good compromise.
In this study, samples were introduced into the capillary electrokinetically. The injection time directly affected the amount of sampling, which affected the peak height and the peak shape. The effect of the injection time on CE separation was investigated by changing the sampling time from 2 to 10 sec in increments of 2 sec at an injection voltage of 15 kV. It was found that both the peak current and the peak width increased with increasing the sampling time. When the injection time exceeded 6 sec, the peak current leveled off and peak broadening became more severe. In this experiment, 6 sec (at 15 kV) was selected as the optimum injection time, considering the separation and sensitivity.
Through the above-mentioned experiments, the optimum conditions for determining paeonol, p-hydroxyacetophenone and 2,4-hydroxyacetophenone were acquired. The typical electropherogram for a mixture containing 0.25 mM PN, PHA and DHA is shown in Fig. 4. Baseline separation for the three phenolic acetophenone could be achieved within 10 min.
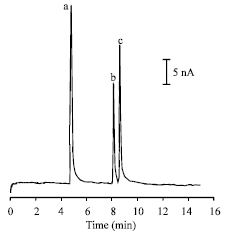 | |
| Fig. 4: | Electropherogram for a mixture containing 0.25 mM of paeonol (a), p-hydroxyacetophenone (b) and 2,4-hydroxyacetophenone (c). Working potential: +0.90 V (vs. SCE); other conditions were as in Fig. 2 |
Precision, Linearity and Detection Limits
The precision of the peak current was examined from a series of 7 repetitive injections of a sample mixture containing 0.25 mM PN, PHA and DHA under the optimum conditions. Reproducible signals were obtained with RSD of 2.2% (PN), 3.5% (PHA) and 2.7% (DHA) for the peak currents. Such good repeatability reflects the reduced surface fouling of the carbon electrode, indicating that this approach is suitable for the analysis of real samples.
A series of the standard mixture solutions of PN, PHA and DHA with concentrations ranging from 1 μM to 1 mM were tested to determine the linearity at the carbon disc electrode in this method. The carbon electrode detector offers a well-defined concentration dependence. The results of a regression analysis on the calibration curves and the detection limits are presented in Table 1. The detection limits
| Table 1: | Results of regression analysis on calibration and detection limits a) |
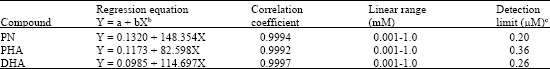 | |
| a Working potential was +0.90 V (vs. SCE). Other conditions were as in Fig. 2, bWhere the Y and X are the peak current (nA) and concentration of the analytes (mM), respectively, cThe detection limits correspond to concentrations giving a signal-to-noise ratio of 3 were evaluated based on a signal-to-noise ratio of 3. | |
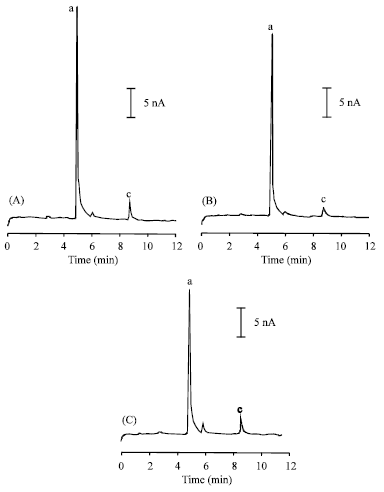 | |
| Fig. 5: | Typical electropherograms for the diluted extracts from three samples of Radix Cynanchi Paniculati (A, Sample 1, B, Sample 2, C and Sample 3). Working potential: +0.90 V (vs. SCE); other conditions were as in Fig. 2. Peak identification: (a) paeonol, (b) 2,4-hydroxyacetophenone |
| Table 2: | Determined contents of paeonol (PN), p-hydroxyacetophenone (PHA) and 2,4-hydroxyacetophenone (PHA) in Radix Cynanchi Paniculati (n = 3, mg g‾1) a) |
 | |
| a Working potential was +0.90 V (vs. SCE). Other conditions were same as in Fig. 2, bThe data in the brackets are the RSDs (%), cNF denotes not found | |
The calibration curves exhibit a satisfactory linear behavior over the concentration range of 3 orders of magnitude with the detection limits of 0.20 μM for PN, 0.36 μM for PHA and 0.26 μM for DHA, respectively.
Sample Analysis and Recovery
Under the optimum conditions, CE-ECD was applied for the determination of PN, PHA and DHA in traditional Chinese medicine, Radix Cynanchi Paniculati. The typical electropherograms for the diluted extracts from three plant samples are shown in Fig. 5A-C. Peak identification was performed by the standard addition method. The assay results are summarized in Table 2. p-Hydroxyacetophenone has not been found in all the three samples of Radix Cynanchi Paniculati. By comparing the electropherograms carefully, it was found that all samples had similar constituents based on the migration times of the peaks. Because both paeonol and 2,4-hydroxyacetophenone were found presented in all the three samples, they can be defined as characteristic markers in the fingerprint of Radix Cynanchi Paniculati under the selected conditions. The determined contents of paeonol in Radix Cynanchi Paniculati are well in agreement with previous reports (Pharmacopoeia of People’s Republic of China, 2005; Zhan and Li, 2004; Yu, 2005; Deng et al., 2006; Guo et al., 1996). In order to prevent the degradation of the analytes, the stock solution and the extracts of the plant samples were diluted using the alkaline running buffer just prior to CE analysis.
Recovery experiments were performed by adding accurate amounts of PN, PHA and DHA to the diluted extract of Radix Cynanchi Paniculati in the running buffer. Subsequently, the standard-spiked sample solution was analyzed under the optimum conditions. The average recoveries and the corresponding RSDs were 98.1 and 3.5% for PN, 97.5 and 2.7% for PHA and 98.7 and 3.4% for DHA, respectively (n = 3). The results demonstrated that this method had both high recovery and good precision for the three phenolic acetophonones.
CONCLUSIONS
For the first time, CE-ECD was employed for the determination of paeonol, p-hydroxyacetophenone and 2,4-hydroxyacetophenone in Radix Cynanchi Paniculati. It is characterized by its higher resolution and sensitivity, lower expense of operation and less amount of sample. The main advantage of CE as an analytical technique for the analysis of plant samples is that capillary is much easier to clean. Because paeonol, p-hydroxyacetophenone and 2,4-hydroxyacetophenone are directly detected on carbon electrode, samples do not need derivatization before determination. It is concluded that CE-ECD is an efficient approach for the constituent and fingerprint study of plant drugs due to its special attributes.
ACKNOWLEDGMENTS
This research was financially supported by the National Science Foundation of China (NSFC 20405002 and 20675017) and Shanghai Science Committee (051107089).
REFERENCES
- Baldwin, R.P., 2000. Recent Advances in electrochemical detection in capillary electrophoresis. Electrophoresis, 21: 4017-4028.
Direct Link - Chen, G., X.H. Han, L.Y. Zhang and J.N. Ye, 2002. Determination of purine and pyrimidine bases in DNA by micellar electrokinetic capillary chromatography with electrochemical detection. J. Chromatogr. A, 954: 267-276.
Direct Link - Chen, G., L.Y. Zhang, X.L. Wu and J.N. Ye, 2005. Determination of mannitol and three sugars in Ligustrum lucidum Ait. by capillary electrophoresis with electrochemical detection. Anal. Chim. Acta, 530: 15-21.
Direct Link - Chou, T.C., 2003. Anti-inflammatory and analgesic effects of paeonol in carrageenan-evoked thermal hyperalgesia. Br. J. Pharm., 139: 1146-1152.
Direct Link - Chu, Q.C., L. Fu, Y.Q. Guan and J.N. Ye, 2004. Determination and differentiation of Flos chrysanthemum based on characteristic electrochemical profiles by capillary electrophoresis with electrochemical detection. J. Agric. Food Chem., 52: 7828-7833.
Direct Link - Deng, C.H., N. Yao, B. Wang and X.M. Zhang, 2006. Development of microwave-assisted extraction followed by headspace single-drop microextraction for fast determination of paeonol in traditional Chinese medicines. J. Chromatogr. A, 1103: 15-21.
CrossRef - Fliser, D., S. Wittke and H. Mischak, 2005. Capillary electrophoresis coupled to mass spectrometry for clinical diagnostic purposes. Electrophoresis, 26: 2708-2716.
Direct Link - Guan, Y.Q., T. Wu, M. Lin and J.N. Ye, 2006. Determination of pharmacologically active ingredients in sweet potato (Ipomoea batatas L.) by capillary electrophoresis with electrochemical detection. J. Agric. Food Chem., 54: 24-28.
Direct Link - Guo, L.B., J.J. Liang and Q.Y. Yang, 1996. Determination of paeonol in the root of Cynanchum paniculatum (Bge.) Kitag by gas chromatography. China J. Chin. Materia Med., 21: 484-485, 511.
Direct Link - Holland, L.A. and A.M. Leigh, 2002. Amperometric and voltammetric detection for capillary electrophoresis. Electrophoresis, 23: 3649-3658.
Direct Link - Huck, C.W., G. Stecher, H. Scherz and G. Bonn, 2005. Analysis of drugs, natural and bioactive compounds containing phenolic groups by capillary electrophoresis coupled to mass spectrometry. Electrophoresis, 26: 1319-1333.
Direct Link - Kim, S.H., S.A. Kim, M.K. Park, S.H. Kim and Y.D. Park et al., 2004. Paeonol inhibits anaphylactic reaction by regulating histamine and TNF-alpha. Int. Immunopharmacol., 4: 279-287.
CrossRef - Lin, C.C., Y.T. Li and S.H. Chen, 2003. Recent progress in pharmacokinetic applications of capillary electrophoresis. Electrophoresis, 24: 4106-4115.
Direct Link - Peng, Y.Y., J.N. Ye and J.L. Kong, 2005. Determination of phenolic compounds in Perilla frutescens L. by capillary electrophoresis with electrochemical detection. J. Agric. Food Chem., 53: 8141-8147.
Direct Link - Piergiovanni, A.R. and G. Taranto, 2005. Simple and rapid method for the differentiation of Lens culinaris Medik. from false lentil species. J. Agric. Food Chem., 53: 6593-6597.
Direct Link