
G.S. Nagananda
Department of Plant Biotechnology, Genohelix Biolabs-A Division of CASB, Jain University, 127/3, Bull Temple Road, Chamrajpet, Bangalore-560019, India
Nalini Satishchandra
Shri Bhagawan Mahaveer Jain College, Jain University, 91/2, Dr. A N Krishna Rao Road, V V Puram, Bangalore-560004, India
S. Rajath
Department of Plant Biotechnology, Genohelix Biolabs-A Division of CASB, Jain University, 127/3, Bull Temple Road, Chamrajpet, Bangalore-560019, India
Journal of Medical Sciences
Year: 2013 | Volume: 13 | Issue: 6 | Page No.: 401-409







ABSTRACT
Flickingeria nodosa (Dalz.) Seidenf is an epiphytic orchid with pseudobulbs which has medicinal importance in traditional system of medicine. In charak samhita it is known as Jeewanti which means life promoter. Based on its importance the present work was designed to evaluate its phytochemical constituents and free radical scavenging activity of pseudobulb cold and hot successive extracts. The phytochemical secondary metabolite screening of extracts revealed the presence of alkaloids, flavonoids, phenols, phytosterols and total antioxidant components. Based on the quantitative estimation studies it revealed that extracts have a good amount of secondary metabolites. The cold and hot successive extracts were subjected to free radical scavenging activity on DPPH, ABTS radical cation decolorization assay, Hydroxyl radical scavenging activity and Nitric oxide scavenging activity. The result revealed that the highest DPPH scavenging and ABTS scavenging activity was seen in the cold acetone extract, hydroxyl radical scavenging activity was more with hot chloroform extract and nitric oxide scavenging activity was more with cold water extracts. The plant can be a source material to herbal drug industry since it is a reservoir of phytochemical components that can be used for the development of therapeutic phytomedicine for the therapy and treatments.
PDF Abstract XML References Citation
Received: April 01, 2013;
Accepted: April 18, 2013;
Published: July 04, 2013
How to cite this article
G.S. Nagananda, Nalini Satishchandra and S. Rajath, 2013. Phytochemical Evaluation and in vitro Free Radical Scavenging Activity
of Cold and Hot Successive Pseudobulb Extracts of Medicinally Important Orchid
Flickingeria nodosa (Dalz.) Seidenf. Journal of Medical Sciences, 13: 401-409.
DOI: 10.3923/jms.2013.401.409
URL: https://scialert.net/abstract/?doi=jms.2013.401.409
DOI: 10.3923/jms.2013.401.409
URL: https://scialert.net/abstract/?doi=jms.2013.401.409
INTRODUCTION
Orchids, generally considered as the most beautiful creation of the Mother Nature, comprise a group of flowering plants and are known to the natives since ancient times. The use of orchids in the system of indigenous medicine dates back to vedic period. The therapeutic use are in practice in different traditional methods of medicines like Ayurveda, Siddha and Unani (Rao, 1998). Traditional therapeutic uses of a number of orchids and their phytochemical constituents like alkaloids, flavonoids, terpenes, carbohydrates and glycosides have been documented in ethanobotanical literatures (Singh and Duggal, 2009). There are unmistakable references of orchids as medicinal constituents in ancient Sanskrit literature like “Charaka Samhita”, “Nighantus” and “Amarakosha” by Charaka, Sushruta and Vagbhata, respectively (Hegde, 1984). In Ayurvedic system, “Ashtawarga”, a group of eight drugs, is used for preparation of tonics such as “Chyavanprash” and consists of four orchid species out of which Flickingeria nodosa is also one among them (Kaushik, 1983). They are called as “Purusharathna” in Kannada. In Charak Samhita, it is known as Jeewanti means the life promoter (Rao, 1998). In ayurvedic and traditional medicines it is believed that it can cure tridosha (Vatta, Pittha and Kapha; Rao, 1998).
Antioxidants are the compounds that can delay or inhibit the oxidation of biomolecules by regulation of oxidative chain reactions (Arya and Yadav, 2011). The antioxidative effects are mainly due to secondary metabolite components like phenolic components. The phenolic compounds have antioxidative activity mainly due to their redox potential, which plays an important role in absorbing and neutralizing free radicals (Butkhup and Samappito, 2011; Coulidiati et al., 2011). Many researchers have investigated powerful antioxidants from natural herbal sources to prevent the reactive oxygen species related disorders in human as well as replace the synthetic compounds (Alam et al., 2012). So the present investigation was started with an aim to evaluate the phytochemical components and in vitro free radical scavenging activity of cold and hot successive pseudobulb extracts of medicinally important orchid Flickingeria nodosa (Dalz.) Seidenf.
MATERIALS AND METHODS
Preparation of the plant material: The pseudobulb stem plant material was collected from the natural habitat and rinsed with distilled water to remove the runaway dust particles. The water was removed by blotting over a filter paper. The plant materials were shade dried and powdered. Ten grams of powdered plant materials was weighed, taken in a muslin cloth and made into packets. The packets were used for the successive extraction by using 5 solvents namely petroleum ether, chloroform, acetone, ethanol and water.
Cold successive extraction: Ten grams of powdered plant material made into packet was soaked in petroleum ether and incubated at 25°C on an orbital shaker at 100 rpm for 24 h. The solvent was decanted into collection bottle and the fresh solvent was added repeatedly till all the plant metabolites were leached out. Then the packet containing the plant material was dried and the extraction was carried out with next successive solvents. The successive extracts were dried by using rotary vacuum evaporator. The dried extracts were used for further studies.
Hot successive extraction: Ten grams of powdered plant material made into packet was placed in the soxhlet extraction apparatus basket which is a vessel with perforated sides and bottom so that liquid can fall through it. When gentle heat is applied to the main flask, the solvent begins to evaporate and the solvent vapours reach the cold condenser at the top of the flask and begin to liquefy on the condenser. The re-condensed solvent on the sides of the condenser begins flowing down the sides of the condenser and begins dripping off of drip points on the end of the condenser. This solvent drips into the top of the soxhlet basket. The solvent flows through the basket and out of the holes in the bottom of the basket carrying the extract with it into the bottom of the flask. The extract laden solvent falling from the soxhlet basket is dark in color and as it becomes clearer, one can know that the plant material is leached out and the process is finished. Then the packet containing the plant material was dried and the extraction was carried out with next successive solvents. The successive extracts were dried by using rotary vacuum evaporator. The dried extracts were used for further studies.
Qualitative phytochemical screening: The different qualitative chemical tests were performed for establishing phytochemical profile of ten extracts obtained from cold and soxhlet successive extractions. The following tests were performed on all the extracts to detect various phytoconstituents present in them (Raaman, 2006).
Detection of alkaloids: The dried extracts were dissolved in few mL of 0.1N HCl and used for all the following tests.
Mayer’s test (Evans, 1997): One milliliter of sample was taken and 1-2 drops of Mayer’s reagent was added from the sides of the test tubes. The appearance of white precipitate indicates the presence of alkaloids in the sample.
Wagner’s test (Wagner, 1993): One milliliter of sample was taken and 2-3 drops of Wagner’s reagent was added from the sides of the test tubes. The appearance of reddish-brown precipitate indicates the presence of alkaloids in the sample.
Hager’s test (Wagner et al., 1996): One milliliter of sample was taken and 1-2 mL of Hager’s reagent was added into the test tubes. The appearance of yellow precipitate indicates the presence of alkaloids in the sample.
Dragendroff’s test (Waldi, 1965): One milliliter of sample was taken and 1-2 mL of Dragendroff’s reagent was added into the test tubes. The appearance of yellow precipitate indicates the presence of alkaloids in the sample.
Detection of Saponins by foam test (Kokate, 1999): Fifty milligram of extract was diluted with distilled water and volume was made upto 20 mL in a measuring cylinder. It was shaken well for 15 min. The appearances of layer of foam of about 2 cm indicate the presence of saponins.
Detection of phytosterols by Liebermann-Burchards method (Finar, 1986): Sample was dissolved in 2 mL of acetic anhydride and 1-2 drops of concentrated H2SO4 was added from the sides of the test tube. The array of change in colours from purple to green indicates the presence of phytosterols.
Detection of phenolic compounds
Ferric chloride (Mace, 1963): The extract was dissolved in 5 mL of distilled water and few drops of neutral 5% FeCl3 solution were added. The appearance of dark green colour indicates the presence of phenols.
Gelatin (Evans, 1997): The extract was dissolved in 5 mL of distilled water and 2 mL of 1% gelatin was added. Appearance of white precipitate indicates the presence of phenols.
Folin-Ciocalteau reagent (Sadasivam and Manickam, 1997): The extract was dissolved in 3 mL of water, 0.5 mL of Folin-Ciocalteau reagent was added and incubated for 3 min and 2 mL of 20% Na2CO3 solution was added. Appearance of blue coloured complex indicates the presence of phenols.
Detection of flavonoids by aluminium chloride method (Zhishen et al., 1999): A 0.5 mL of extract was mixed with 2 mL of distilled water and subsequently with 0.15 mL of 5% NaNO2 solution. After 6 min, 0.15 mL of a 10% AlCl3 solution was added and allowed to stand for 6 min, then 2 mL of 4% NaOH solution was added to the mixture. Immediately distilled water was added and then the mixture was incubated at RT for 15 min. Pink colour indicates the presence of flavonoids.
Detection of Glycosides by Borntrager’s test (Evans, 1997): Fifty milligram of extract was hydrolyzed with concentrated hydrochloric acid for 2 h on a water bath and filtered. To 2 mL of filtered hydrolysate, 3 mL of chloroform’ was added and shaken, chloroform layer is separated and 10% ammonia solution was added to it. Pink colour indicates the presence of glycosides.
Estimation of alkaloids (Sreevidja and Mehrotra, 2003): The calibration curve was obtained with Bismuth nitrate pentahydrate stock solution. Series dilutions of the stock solution were made by pipetting out from 200 to 1000 μL and the volume was made upto 1 mL with distilled water, 5 mL of 3% thiourea solution was added to it. The absorbance value of the yellow solution was measured at 435 nm against colorless reagent blanks.
A 5 mL amount of the extract/solution was taken and the pH was maintained at 1.2-2 with dilute HCl. A 2 mL amount of Dragendroff’s Reagent (DR) was added to it and the precipitate formed was centrifuged. The centrifugate was checked for complete precipitation by adding DR. After centrifugation, the centrifugate was decanted completely and meticulously. The precipitate was further washed with alcohol. The filtrate was discarded and the residue was then treated with 2 mL of 1% disodium sulfide solution. The brownish black precipitate formed was then centrifuged. Completion of precipitation was checked by adding 2 drops of disodium sulfide. The residue was dissolved in 2 mL concentrated nitric acid. This solution was diluted to 10 mL in a standard flask with distilled water; 1 mL was then pipetted out and 5 mL of 3% thiourea solution was added to it and absorbance was measured at 435 nm. Two milliliter of nitric acid was diluted to 10 mL in a standard flask with distilled water; 1 mL was then pipetted out and 5 mL thiourea solution was added to it and which serves as a blank.
Estimation of phytosterols by libermann-burchard method (Finar, 1986): Prepare standard cholesterol solutions (1 mg mL-1) in chloroform. Pipette out different aliquots of standard cholesterol ranging from 0.2-1.0 mL in test tubes. Add 2 mL of acetic anhydride and 1-2 drops of concentrated H2SO4. Make up the volume to equal quantity using chloroform. Incubate in dark for 15 min. Prepare the blank with 1 mL chloroform and 2 mL acetic anhydride. Sample was dissolved in 1 mL of chloroform and 2 mL of acetic anhydride was added to it, absorbance was measured at 640 nm.
Estimation of total flavonoid content
Aluminium chloride method (Zhishen et al., 1999): Different aliquots (0.2-1 mL) of standard Quercetin (100 μg mL-1) solution were taken in a series of test tubes. Five hundred micro liter of extract was mixed with 2 mL of distilled water and subsequently with 0.15 mL of 5% NaNO2 solution. After 6 min, 0.15 mL of a 10% AlCl3 solution was added and allowed to stand for 6min and then 2 mL of 4% NaOH solution was added to the mixture. Immediately distilled water was added to bring the final volume of 5 mL and then the mixture was thoroughly mixed and allowed to stand at RT for 15 min. Absorbance of the mixture was determined at 510 nm.
Estimation of total phenols (Sadasivam and Manickam, 1997): The extracts were dissolved in 5 mL of distilled water. Different aliquots (0.2-1 mL) of standard Catechol (50 μg mL-1) solution were taken in a series of test tubes. The volume in each tube was made up to 5 mL with water. 0.5 mL of Folin-Ciocalteau reagent was then added to each test tube and mixed well. After 3 min, 2 mL of 20% Na2CO3 solution was added to each tube and mixed thoroughly. The tubes were kept in the boiling water for exactly one minute and then cooled. The absorbance against a reagent blank was taken at 650 nm.
Total antioxidant capacity
Phosphomolybdenum assay (Prieto et al., 1999): One hundred micro liter of the extract dissolved in DMSO was mixed in the eppendorf tube with 1 mL of total antioxidant reagent (0.6 M sulphuric acid, 28 mM sodium phosphate and 4 mM ammonium molybdate). Different aliquots (20-100 μg mL-1) of standard ascorbic acid were taken into series of eppendorf tubes and the volume was made up to 0.1 mL with DMSO and 1 mL of total antioxidant reagent was added. The eppendorf tubes were capped and incubated in a thermal block at 95°C for 90 min. Cooled to room temperature and the absorbance measured at 695 nm against black.
To evaluate the antioxidant properties of different phytochemical extracts: Free radical scavenging activity on 1, 1-diphenyl-2-picrylhydrazyl (DPPH) (Blois, 1958) Standard ascorbic acid Sample extracts (1 mg mL-1) at various concentration (10-50 μg mL-1) were taken and the volume was adjusted to 100 μL with methanol. Five milliliter of a 0.1 mM methanolic solution of DPPH was added and shaken vigorously. The tubes were allowed to stand for 20 min at 27°C. The absorbance of the sample was measured at 517 nm. Methanol serves as a blank and prepared DPPH serves as a control and the experiment was performed in triplicate. Radical scavenging activity was expressed as the inhibition percentage of free radical by the sample and was calculated using the formula:
ABTS (2, 2-azinobis (3-ethylbenzothiazoline-6-sulfonic acid)) radical cation decolorization assay (Re et al., 1999): ABTS radical cation (ABTS+) was produced by reacting ABTS (7 mM) with 2.45 mM ammonium persulfate and the mixture was allowed to stand in dark at room temperature for 12-16 h before use. Standard ascorbic acid and sample extracts (10 mg mL-1) at various concentrations (200-1000 μg mL-1) were taken and the volume was adjusted to 500 μL with DMSO and 500 μL of DMSO serves as blank. Three hundred micro liter of ABTS solution was added; the final volume was made up with ethanol to make 1 mL and incubated in dark for 30 min at RT. The absorbance was read at 745 nm and the experiment was performed in triplicate. Radical cation decolorization activity was expressed as the inhibition percentage of cations by the sample and was calculated using the formula:
Nitric oxide scavenging assay (Sreejayan and Rao, 1997): Different concentrations of standard ascorbic acid and sample extract (200-1000 μg mL-1) dissolved in DMSO were taken and the volume was adjusted to 1.5 mL with sodium nitroprusside (5 mM) in phosphate buffer saline and incubated at 25°C for 30 min. After 30 min, it was diluted with 1.5 mL of Griess reagent (1% sulphanilamide, 2% orthophosphoric acid and 0.1% napthyl ethylene diamine dihydrochloride). The absorbance of the chromphore formed during diazotization of the nitrite with suplhanilamide and subsequent coupling with naphlethylene diamine was measured at 546 nm along with a control. The percentage inhibition of nitric oxide generated was measured by comparing the absorbance values of control and test sample using following formula:
Hydroxyl radical scavenging activity (Klein et al., 1991): The scavenging activity of extracts on hydroxyl radical was measured according to the method of Klein et al. (1991). Various concentrations (20-100 μg mL-1) of extracts (1 mg mL-1) and standard ascorbic acid (1 mg mL-1) were added with 1 mL of iron-EDTA solution (0.13% ferrous ammonium sulfate and 0.26% EDTA), 0.5 mL of EDTA solution (0.018%) and 1.0 mL of diemthyl sulphoxided (DMSO) (0.85% v/v in 0.1 M phosphate buffer, pH 7.4). The reaction was initiated by adding 0.5 mL of ascorbic acid (0.22%) and incubated at 80-90°C for 15 min in a water bath. After incubation the reaction was terminated by the addition of 1 mL of ice-cold TCA (17.5% w/v). Three milliliters of Nash reagent (75 g of ammonium acetate, 3 mL of glacial acetic acid and 2 mL of acetyl acetone were mixed and raised to 1 liter with distilled water) was added and left at room temperature for 15 min. The reaction mixture without sample was used as control. The intensity of the color formed was measured spectroscopically at 412 nm against reagent blank. The% hydroxyl radical scavenging activity (HRSA) is calculated by the following formula:
IC50 value: IC50 values (concentration of sample required to scavenge 50% of free radicals) were calculated from the regression equation, prepared from the concentration of the samples and percentage inhibition of free radical formation. Ascorbic acid was used as positive control and all tests were carried out in triplicate.
Statistical analysis: The experiments were set up in a completely randomized design. All values obtained from the mean replicates were averaged. The data were analyzed in relation to the variance and presented as mean±standard error (SE). Analysis of variance was conducted by two way ANOVA and the mean were compared by Tukey HSD test. All statistical analysis was performed at 1% significance level using IBM SPSS Statistics (version 20) by IBM.
RESULTS
Qualitative phytochemical screening: The different qualitative chemical tests were performed for establishing phytochemical profile of ten extracts obtained from cold and soxhlet successive extractions. Phytochemical screening was performed for all the extracts which revealed the presence of alkaloids, flavonoids, phenols and phytosterols in different extracts (Table 1).
Quantitative estimation of phytochemicals: The quantitative estimations of the phytochemicals, which were qualitatively detected in the pseudobulb stem revealed the presence of high alkaloid content (50.5 μg mL-1) in hot chloroform extract (Fig. 1). The presence of high phenol content (225.84 μg mL-1) was recorded in cold acetone extracts (Fig. 2). High content of phytosterols (24 μg mL-1) was recorded in hot acetone extract (Fig. 3).
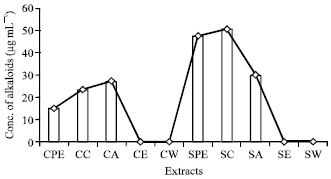 | |
| Fig. 1: | Concentration of alkaloids (μg mL-1) present in different extracts |
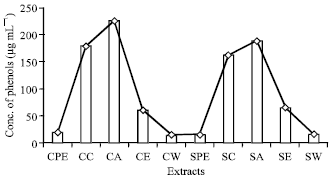 | |
| Fig. 2: | Concentration of phenols (μg mL-1) present in different extracts |
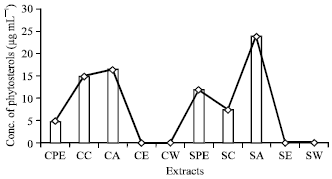 | |
| Fig. 3: | Concentration of phytosterols (μg mL-1) present in different extracts |
| Table 1: | Phytochemical screening of ten extracts of Flickingeria nodosa |
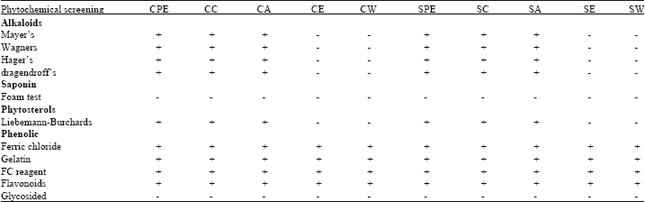 | |
| C: Cold extract, S: Soxhlet hot extract, PE: Petroleum ether, C: Chloroform, A: Acetone, E: Ethanol ,W: Aqueous | |
| Table 2: | Quantitative estimation of Phytochemical |
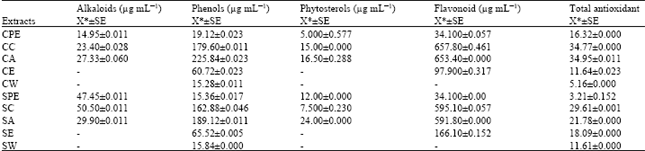 | |
| *Mean of 3 replications, SE: Standard error | |
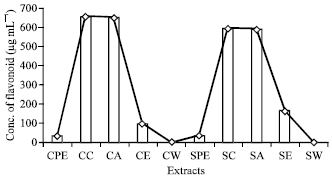 | |
| Fig. 4: | Concentration of Flavonoid (μg mL-1) present in different extracts |
High content of flavonoids (657.80 μg mL-1) was recorded in cold chloroform extract (Fig. 4). High total antioxidant content (34.95 μg mL-1) was recorded in cold acetone extracts (Fig. 5). The present study reveals that the cold extraction is more effective for phenols, flavonoids and total antoxidants while hot extraction is more effective for alkaloids and phytosterols (Table 2).
Free radical scavenging activity assay
DPPH free radical scavenging activity assay: Free radical scavenging potential of extracts at different concentrations was tested by DPPH method.
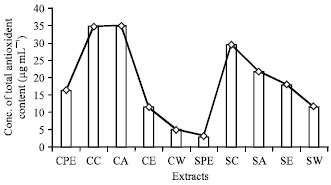 | |
| Fig. 5: | Concentration of total antioxidant content (μg mL-1) present in different extracts |
Highest percentage of scavenging activity and IC50 values were found to be 43.68% and 46.49 μg mL-1 respectively for cold acetone extract. The standard ascorbic acid showed the percentage of scavenging activity and IC50 values as 60.878% and 21.04 μg mL-1 (Table 3-4) (Fig. 6).
ABTS radical scavenging assay: Radical cation decolorization activity of extracts at different concentrations was tested by ABTS method. Highest percentage of scavenging activity and IC50 values were found to be 73.11% and 12.7 μg mL-1 respectively for cold acetone extract.
| Table 3: | Percentage of scavenging activity of different extracts against different assays |
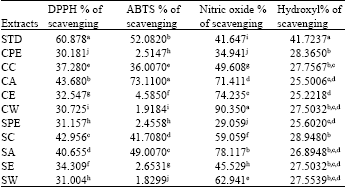 | |
| Mean of 15 replicate. Mean values with different superscripts (a,b,c,d,e,f,g,h,I,j and k) differ significantly at p<0.01 by Tukey (HSD) test | |
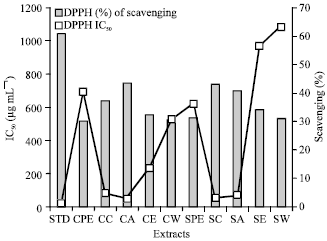 | |
| Fig. 6: | DPPH scavenging activity of different extracts and its IC50 (μg mL-1) |
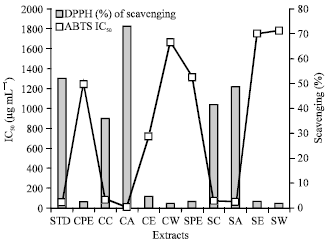 | |
| Fig. 7: | ABTS scavenging activity of different extracts and its IC50 (μg mL-1) |
The standard ascorbic acid showed the percentage of scavenging activity and IC50 values as 52.082% and 55.66 μg mL-1 (Table 3-4) (Fig. 7).
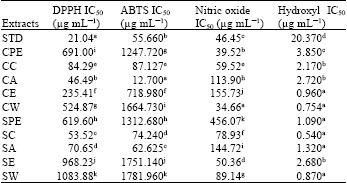
| Table 4: | IC50 value of 10 extracts different antioxidant assays |
 | |
| Values with different superscripts (a,b,c,d,e,f,g,h,I,j and k) differ significantly | |
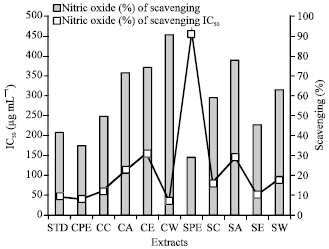 | |
| Fig. 8: | Nitric oxide radical scavenging activity of different extracts and its IC50 (μg mL-1) |
Nitric oxide scavenging assay: The extracts at different concentrations were tested for nitric oxide radical scavenging activity. Highest percentage of activity and IC50 values were found to be 90.352% and 34.66 μg mL-1, respectively for cold water extract. The standard ascorbic acid showed the percentage of scavenging activity and IC50 values as 41.647% and 46.45 μg mL-1 (Table 3-4) (Fig. 8).
Hydroxyl radical scavenging activity assay: The extracts at different concentrations were tested for hydroxyl radical scavenging activity. Highest percentage of activity and IC50 values were found to be 28.9480% and 0.54 μg mL-1, respectively for hot chloroform extract. The standard ascorbic acid showed the percentage of scavenging activity and IC50 values as 41.7237% and 20.37 μg mL-1 for ascorbic acid (Table 3-4) (Fig. 9).
DISCUSSION
Based on the ethnobotanical literature, Flickingeria nodosa has a great medicinal importance for having high amount of secondary metabolites (Kaushik, 1983).
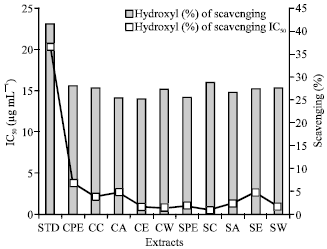 | |
| Fig. 9: | Hydroxyl radical scavenging activity of different extracts and its IC50 (μg mL-1) |
In the present investigation, hot and cold successive extraction was carried with different solvents (Petroleum ether, chloroform, acetone, ethanol and water), revealing the presence of alkaloids, flavonoids and phenols, which agrees with the earlier findings of Chhajed et al.(2008). The extracts obtained from the present study revealed the presence of phytosterols, which was not reported earlier. The cold extraction was more effective for extracting phenols and flavonoids, whereas hot extraction was more effective for alkaloids and phytosterol extraction.
Free radicals are produced under certain environmental conditions and during normal cellular functions in the body. These molecules are missing an electron, giving them an electric charge. To neutralize this charge, free radicals try to withdraw an electron from, or donate an electron to, a neighbouring molecule. The newly created free radical, in turn, looks out for another molecule and withdraws or donates an electron, setting off a chain reaction that can damage hundreds of molecules. Antioxidants halt this chain reaction. Some antioxidants are themselves free radicals, donating electrons to stabilize and neutralize the dangerous free radicals. Other antioxidants work against the molecules that form free radicals, destroying them before they can begin the domino effect that leads to oxidative damage (Matill, 1947). In the present investigation, total antioxidant activity was determined with different antioxidant assays. High amount of scavenging activity with an evident IC50 was seen with the cold acetone and cold water extracts when compared to that of hot extracts (Chhajed et al., 2008). As compared to the hot extracts, cold extracts have given a better activity. This is because the bioactive component present in the extracts might be thermolabile, which might lose its activity when extracted under heat. In the present investigation, high amount of DPPH scavenging activity, ABTS scavenging activity and nitric oxide scavenging activity with an evident IC50 value was observed. The DPPH involves in their hydrogen-donating ability, ABTS chemistry involves direct generation of ABTS radical mono cation with no involvement of any intermediary radical and nitric oxide involves in reduction of nitrogen-free radicals. Hence, the more scavenging activity directly relates their antioxidant capacity. Thus the cold extracts are more reducing agent for reactive oxygen species and nitric oxide-free radical. The cold acetone extract has given good scavenging activity against DPPH and ABTS radical and cold water extract has given good scavenging activity against nitric oxide radical. These extracts have shown high antioxidant activity due to the presence of phenolic compounds which agrees with the findings of Materska and Perucka, (2005). The percentage of hydroxyl radical scavenging activity does not show significant difference in result for different extracts. The IC50 value for hot chloroform extract was found to be the best as compared to other extracts.
CONCLUSION
The plant can be a source material to herbal drug industry since it is a reservoir of phytochemical components that can be used for the development of therapeutic phytomedicine for the therapy and treatments.
ACKNOWLEDGMENTS
We acknowledge Dr. R. Chenraj Jain, President, Jain University Trust., Dr. N Sundararajan, Vice Chancellor, Jain University., Prof. K.S. Shantamani, Chief Mentor, Jain University and Dr. S Sundara Rajan, Director, CASB-Jain University, Bangalore for providing financial assistance, the necessary laboratory facilities and support. We also thank the staff at Genohelix Biolabs for technical support.
REFERENCES
- Singh, A. and S. Duggal, 2009. Medicinal orchids: An overview. Ethnobotanical. Leaflets, 13: 351-363.
Direct Link - Blois, M.S., 1958. Antioxidant determinations by the use of a stable free radical. Nature, 181: 1199-1200.
CrossRefDirect Link - Butkhup, L. and S. Samappito, 2011. In vitro free radical scavenging and antimicrobial activity of some selected Thai medicinal plants. Res. J. Med. Plant, 5: 254-265.
CrossRefDirect Link - Klein, S.M., G. Cohen and A.I. Cederbaum, 1981. Production of formaldehyde during metabolism of dimethyl sulfoxide by hydroxyl radical generating systems. Biochemistry, 20: 6006-6012.
CrossRefPubMedDirect Link - Alam, B.M., M.S. Rahman, M. Hasan, M.M. Khan, K. Nahar and S. Sultana, 2012. Antinociceptive and Antioxidant activities of the Dillenia indica bark. Int. J. Pharm., 8: 243-251.
CrossRefDirect Link - Materska, M. and I. Perucka, 2005. Antioxidant activity of the main phenolic compounds isolated from hot pepper fruit (Capsicum annuum L.). J. Agric. Food Chem., 53: 1750-1756.
CrossRefPubMedDirect Link - Prieto, P., M. Pineda and M. Aguilar, 1999. Spectrophotometric quantitation of antioxidant capacity through the formation of a phosphomolybdenum complex: Specific application to the determination of vitamin E. Anal. Biochem., 269: 337-341.
CrossRefDirect Link - Raaman, N., 2006. Phytochemical Techniques. New India Publishing Agency, New Delhi, India, ISBN: 9788189422301 Pages: 320.
Direct Link - Re, R., N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, 1999. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radical Biol. Med., 26: 1231-1237.
CrossRefDirect Link - Sreejayan and M.N.A. Rao, 1997. Nitric oxide scavenging by curcuminoids. J. Pharm. Pharmacol., 49: 105-107.
CrossRefPubMedDirect Link - Sreevidja, N. and S. Mehrotra, 2003. Spectrophotometric method for estimation of alkaloids precipitable with Dragendorff's reagent in plant materials. J. AOAC, 86: 1124-1127.
PubMedDirect Link - Coulidiati, T.H., H. Millogo-Kone, A. Lamien-Meda, M. Yougbare-Ziebrou, J. Millogo-Rasolodimby and O.G. Nacoulma, 2011. Antioxidant and antibacterial activities of two Combretum species from burkina faso. Res. J. Med. Plant, 5: 42-53.
CrossRefDirect Link - Arya, V. and J.P. Yadav, 2011. Antioxidant properties of the methanol extracts of the leaves, seeds and stem of Cassia occidentalis. Res. J. Med. Plant, 5: 547-556.
CrossRef - Zhishen, J., T. Mengcheng and W. Jianming, 1999. The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals. Food Chem., 64: 555-559.
CrossRefDirect Link