
Amy M. Kwon
Statistics and Advanced Analytics Division, IMS Health Co., Ltd, Seoul, Korea
Journal of Medical Sciences
Year: 2013 | Volume: 13 | Issue: 1 | Page No.: 1-9
DOI: 10.3923/jms.2013.1.9







ABSTRACT
Metabolomics is a relatively young but emerging functional genomics which produces high-dimensional data. Bioinformatics techniques are playing key roles to interpret these high-throughput metabolomics data and it is critical for researchers to understand bioinformatics techniques to enhance its applications and further development in metabolomics. This article briefly overviews the current study approaches and data generation techniques by different analytical platforms in metabolomics based on highly cited articles. With available preprocessing packages, it discusses the bioinformatics techniques commonly used in metabolomics studies including application examples. Besides analytical techniques, it is obvious that advanced approaches or new applications in bioinformatics speed up the development of metabolomics and there are much potential for bioinformatics to be further applied to the metabolomics data regarding the current state of metabolomics studies. It is impossible to single one method out among the techniques, but it should be encouraged applying more than one approach to the data obtained by current platforms and mixed approaches or data-driven approaches may need to be considered to interpreting the results.
PDF Abstract XML References Citation
Received: January 23, 2013;
Accepted: March 17, 2013;
Published: May 06, 2013
How to cite this article
Amy M. Kwon, 2013. Bioinformatics Methods in Metabolomics Studies. Journal of Medical Sciences, 13: 1-9.
DOI: 10.3923/jms.2013.1.9
URL: https://scialert.net/abstract/?doi=jms.2013.1.9
DOI: 10.3923/jms.2013.1.9
URL: https://scialert.net/abstract/?doi=jms.2013.1.9
INTRODUCTION
As a functional genomics, metabolomics has emerged and it has been quickly developing in recent years. Like other functional genomics, the metabolomics generating a huge and complex data, studies the low molecular weight compounds that cover the specific metabolites. It is expected that different metabolites are caused by various phenotypic responses including drug effects, toxicological responses or disease states (Altmaier et al., 2008). For example, it is known that the metabolism changes significantly and the metabolic network re-distributes when normal cells are transformed to malignant ones (Boros et al., 2003). In that case, the metabolomics attempts to identify these overall metabolic changes which potentially impact pathways to a specific biological process due to these transformations. It also attempts to find the key metabolites (Tsugawa et al., 2011) or the most affected metabolic networks which are associated with these transformations (Goodacre et al., 2004). It sometimes aims to assign samples to distinct phenotypic groups according to the metabolic patterns. All those findings can be done by observing the functional relationships in the metabolomics data, but these data are huge and complex. Moreover, the metabolomics data need extensive preprocessing procedures depending upon analytical platforms before the analysis and bioinformatics techniques are essential to find the functional relationships with the transformations in the primary analysis procedure. Thus, it is important to understand primary bioinformatics methods in order to interpret the metabolomics data. This article provides a basic overview of the study approaches in metabolomics and describes the analytical platforms generating metabolomics data. It briefly summarizes the preprocessing procedure including available software packages and reviews the multivariate statistical methods widely used in metabolomics studies.
MAJOR STUDY APPROACHES
The metabolomics is a comprehensive study of whole metabolome under given conditions and the study approaches in metabolomics can be categorized according to these given conditions as well as the study objectives. In general, three major approaches are used in metabolomics (Vladimir, 2006).
| • | Targeted analysis: The targeted analysis is the study approach having a priori knowledge. When the chemical compounds of interest are known, this study approach aims at measuring the concentration of the limited number of metabolites of a particular enzyme system where these chemical compounds are directly affected by perturbation |
| • | Profiling analysis: This study approach has generally no priori knowledge. This study approach globally searches a class of chemical compounds which are associated with specific pathways without any priori knowledge, so, it usually results in high-throughput measurements of much larger number of metabolites. It is appropriate to observe the global metabolic changes in biological system |
| • | Fingerprinting analysis: This study approach pays attention to a unique pattern of the metabolism in a particular cell line or tissue like a snapshot (Allen et al., 2003; Lucio et al., 2010). This approach usually classifies the samples on the basis of provenance of either their biological relevance or origin according to this unique patterns, so most pattern-recognition techniques can be directly applicable to the study |
ANALYTICAL PLATFORMS FOR DATA GENERATION
The different analytical platforms may affect qualities of metabolomics data in terms of their resolutions and sensitivities as well as they generate different data structures. Most common platforms are NMR (Nuclear Magnetic Resonance) (Cheng et al., 1996) and MS (Mass Spectroscopy) (Qiu et al., 2010; Yang et al., 2007). NMR simultaneously quantifies a wide range of compounds in the micromoles range (Viant et al., 2003). Yet, the mixture of the spectra is difficult to separate and the sensitivity is relatively poor to analyze a large number of low abundant metabolites (Vladimir, 2006). MS platforms are separated according to how to analyze the sample as GC (Gas Chromatography), LC (Liquid Chromatography) and CE (Capillary Electrophoresis) (Britz-McKibbin and Terabe, 2002). GC-MS has an advantage to separate volatile metabolites and available libraries are abundant such as 2005 NIST/EPA/NIH Mass Spectral Library and Wiley Registry of Mass Spectral Data. LC-MS has a bigger coverage of molecular masses (Allen et al., 2003) and CE--MS has high resolution with fewer samples in shorter processing time. Recently, PDA (Photodiode Array) or FTIR (Fourier Transform Infrared Spectroscopy) (Kim et al., 2006) are also used. However, it is impossible to single out a superior platform to consider all diverse metabolites. The primary highlights and lowlights are briefly summarized on Table 1.
| Table 1: | Comparison of analytical platforms in metabolomics |
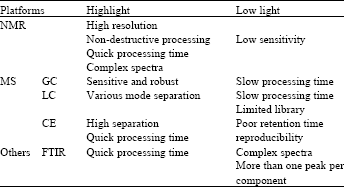 | |
REQUIRED PREPROCESSING
Due to complex characteristics of metabolomics data, the preprocessing procedure is an important procedure in the analysis. This procedure generally includes noise reduction, variation correction, spectrum deconvolution, peak detection, integration and identification (Hansen, 2006; Katajamaa and Oresic, 2007; Scalbert et al., 2009; Spraul et al., 1994). Because different analytical platforms generate different data structure, the preprocessing procedure can be different according to platforms. If the data are obtained by NMR, the preprocessing procedure needs to adjust for variations by measurements. Binning, peak detection, spectra alignment and spectrum deconvolution are usually included in the preprocessing for NMR (Schripsema, 2010; Vogels et al., 1996). If the data are obtained by MS, the raw data need to adjust for background and noise in comparison with the spectral signals. Typically, the preprocessing procedure contains noise reduction, peak detection, spectrum deconvolution, peak integration, chromatogram alignment, component detection and identification, quantification (Katajamaa and Oresic, 2007). There are available softwares for automated data processing. AMDIS (Automated Mass Spectral Deconvolution and Identification System), CODA (Component Detection Algorithm) and WMSM (Windowed Mass Selection Method), Mzmine (Katajamaa et al., 2006), XCMS, XCMS2 are available for data structures from MS platform (Halket et al., 1999; Windig et al., 1996; Fleming et al., 1999; Smith et al., 2006).
BIOINFORMATICS METHODS IN METABOLOMICS
Bioinformatics is also developing as speedy as metabolomics and it plays important roles to interpret the relationship between the metabolomics data and the phenotypic difference. This section separates the bioinformatics techniques which are commonly applied to the metabolomics data into six categories: Correlation based methods, Dimension reduction methods, Regression based methods, Discrimination, Clustering methods and SOM. Notations are denoted as follows. Let X be a matrix of metabolite concentrations whose dimension is nxp where n is the number of samples and p is the number of metabolites. Suppose Y is also a matrix of additional information whose dimension is nxm where n is the number of samples and m is the number of different information.
CORRELATION BASED METHODS
If all metabolites remain at their steady levels, the correlations are assumed not to be seen (Fiehn, 2001). However, if there is any change in the underlying biophysical system, the correlations among the metabolites are observed due to this change. Under this assumption, correlation based methods are usually applied to visualize the correlation between two given metabolites concentrations, or to explore a set of metabolites with stronger correlation before further analysis (Steuer et al., 2003).
Pearson’s correlation coefficient: Pearson's correlation coefficient is a well-known quantity to represent the strength of the association between two numerical variables. The correlation between two given metabolite concentrations of (xi, xj) can be measured as Eq. 1:
| (1) |
where, σxi,xj is a covariance between xi and xj and σxi2 and σxj2 are the variances of xi and xj for i≠ j. The strongly correlated metabolites can be found by choosing the metabolites of |ri,j2| > Ct where Ct is a given threshold.
Partial η2: η2 is a ratio of the variances-the variance explained by the model to the total variance. Altmaier et al. (2008) introduced η2gain to quantify the association between known metabolite concentrations for a specific disease before clustering techniques:
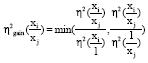 | (2) |
This method allows choosing the metabolite ratios that yield more information than a single metabolite by computing η2 with all possible pairs of metabolites ratios.
DIMENSION REDUCTION METHODS
The dimension reduction is one of the major tasks for high-throughput data such as metabolic profiling data. The purpose of this task is to find a low-dimensional representation capturing the primary contents from high-dimensional metabolites. The linear dimension reduction techniques are well-known and PCA (Principal Component Analysis) (Deo et al., 2010; Halket et al., 1999; Jackson, 1991) and Factor Analysis are commonly used in the metabolomics data.
PCA: PCA is a representative dimension reduction technique which is based on linear projection using second-order statistics. PCA finds the orthogonal linear combinations of metabolite concentrations to maximize the variances. (The variance is proportional to the scale of the variables, standardization of the variables are generally conducted before PCA.) Let T be a set of orthogonal linear combinations using metabolite concentrations, X for j = 1…k and k = p:
| (3) |
Where:
| (4) |
The new set of variables, T is called PCS (Principal Components) and PCA basically finds these uncorrelated PCS from the original metabolite concentrations. The number of PCS is determined with a smaller number than the dimension of the original metabolites concentrations and one way to determine the number of PCS is by Scree plot. Scree plot shows the cumulative proportion of the variance which is explained by each PC and it is customary to find 'elbow' point in the plot. If the elbow plot is located at k+1, then the number of PCS is determined to k. PCA is easy to conduct and strong at visualization, but PCA may not pick up some metabolites whose variances are small in high-throughput data (Blekherman et al., 2011). Most metabolomics studies have adopted PCA either before further analysis or in their analysis (Seierstad et al., 2008; Allen et al., 2003; Bogdanov et al., 2008).
Factor analysis: Factor analysis is also a linear dimension reduction technique based on the second order statistics, but this method is focused on identifying the underlying random quantities which are called ‘factors’. These factors are defined by the relationship between variance matrix and metabolite concentrations and the metabolites are assumed to be dependent upon these factors:
| (5) |
On (5), E(F) =0, E(ε) = 0, Cov(F) = I and Cov(ε) = Φ where, Φ is a diagonal matrix. F indicates a set of k factors where k = p and xj indicates the loading on jth variable. εj is an error term which is independent from F for j = 1,…,p. Factor analysis is sometimes re-parameterized as Eq. 6 after orthogonally rotating factors when the variance is not often factored as Eq. 5. One common rotation technique is ‘varimax’ which constraints to few non-zero loadings (Fodor, 2002). To rotate factors, the new factors F* and the new loadings L* should be defined using orthogonal matrix G as Eq. 6:
| (6) |
The loadings having higher coefficients are often used to identify the biomarker of a specific disease from normal samples in dimension reduction methods (Odunsi et al., 2005). Factor analysis can stand alone as an application technique, but PCA is generally used as an information extraction tool for further analysis.
REGRESSION BASED METHODS
Regression is a technique defining the relationship between two variables. Due to high-dimensional nature of metabolomics data, regression methods are typically performed with dimension reduction techniques in metabolomics and PCR (Principal Components Regression) and PLSR (Partial Least Square Regression) (Wold et al., 2001) are commonly used in metabolomics studies.
PLSR: Once the dimension of metabolites are reduced to k using PCA, regression can be performed using these k principal components as (7) because of xj = αjTT where Ts are principal components and {β0, β1,...,βk} are unknown:
| (7) |
The response variable Y is expressed same as Multiple Linear Regression, the unknown coefficients {β0, β1,.., βk} are estimated by minimizing the least square error and the estimates are expressed as (TTT)(-1)TTY. Although the principal components are a good representation for metabolites, the principal components are chosen based on variances, so they may not be good predictors for Y. This technique has used with magnetic resonance spectroscopy data to elucidate human rectal cancer biopsies and colorectal xenografts and investigate the metabolic change with PCA (Seierstad et al., 2008).
PLSR: PLSR is a popular regression method in metabolomics which adopts PLS technique (Barker and Rayens, 2003; Baumgartner et al., 2011; Bylesjo et al., 2006; Trygg and Wold, 2002). Underlying idea for PLSR is to capture maximizing covariance between T and U from Eq. 8 to estimate Y based on linear model of Y = Xβ+β0:
| (8) |
where, T and U are nxk score matrices for X and Y. P and Q are loading matrices whose dimensions are pxk and mxk. While PCA captures the maximum variances of X, PLS captures the covariance between X and Y. PLSR is good to represent the relationship between additional phenotypic information and metabolites concentrations. NIPALS and SIMPLS are two representative algorithms to conduct PLSR. Both algorithms are known to work accurately, but SIMPLS shows faster convergence than NIPALS. As variants of PLS, MPLS (Multilevel-PLS), OPLS (Orthogonal Projection to Latent Structures) are also applied to metabolomics data (Trygg and Wold, 2002; Van Velzen et al., 2008). In fact, both PCRL and PLSR adopt dimension reduction techniques, so, they are attractive to the researchers who have to deal with a large volume of data, but how to reduce the dimension is different. While PCR focuses on the maximizing variance, PLS focuses on the maximizing covariance. Thus, the performances will vary according to the patterns of the metabolomics data.
DISCRIMINATION METHODS
While regression methods find the relationship between the predictors and continuous outcomes, discrimination methods separate the samples whose outcome variables are discrete according to the discriminant functions based on the predictors. PLS-DA is prevalent in metabolomics profiling studies (Barker and Rayens, 2003; Bylesjo et al., 2006).
PLS-DA: PLS-DA is a variant of PLSR when the outcome variables are discrete. The first procedure for this method is to convert a discrete outcome variable to an extended dummy matrix using either 0 or 1 and the dimension of this dummy matrix is determined by the number of distinct values of the categories in the outcome variable. With this extend dummy matrix, PLS-DA is conducted as the same way to PLSR described above. In metabolomics, the separation the samples into different classes can be interpreted as predicting the unknown origins of the biological samples based on the metabolite concentrations. As a example, this technique has used to separate Parkinson's disease patients from the control samples with blood biomarkers (Bogdanov et al., 2008) and discriminate between idiopathic and LRRK2 Parkinson’s patients (Johansen et al., 2009). It has also used to detect metabolic changes from a profile study with glucose tolerance test (Wopereis et al., 2009). One of the reasons that PLS-DA is prevalently used is, it enables us to separate the specific disease samples from the control samples and to identify possible metabolite biomarkers in profiling data (Barker and Rayens, 2003; Van Velzen et al., 2008). But, PLS-DA is known to show inconsistency in optimization, so the model assessment is also important. The number of misclassification and the area under ROC curve showed efficient results in detecting small differences between groups as diagnostic statistics for PLS-DA (Szymanska et al., 2012).
CLUSTERING METHODS
Clustering analysis is a typical unsupervised learning method. Clustering methods assign samples into clusters based on a given dissimilarity metric. When the number of clusters is unknown, the most prevalent clustering techniques would be Hierarchical clustering method in metabolomics studies. If the number of underlying clusters is known as k, k-Means clustering is prevalently used (Cuperlovic-Culf et al., 2009). The clustering techniques can directly be applied to Non-targeted profiling studies and Fingerprinting studies.
HCA: HCA (Hierarchical Clustering Analysis) has two types. Agglomerative type which is also called ‘bottom-up’ regards each sample as its own cluster at first and recursively merges them by comparing the distance of a pair until all samples are in the same clusters. Oppositely, Divisive type which is called ‘top down’ regards all samples as one cluster and recursively separates samples according to a given distance as one moves down until all samples are split. Regardless of clustering types, hierarchical clustering methods are performed based on distance metrics and linkage criteria, so the clustering results may vary by these two components. Distance metrics are the quantities to represent the distances between samples and widely used metrics are Euclidean distance, Squared Euclidean distance and Mahalonobis distance where j≠k:
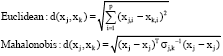 | (9) |
Besides the distance metrics, linkage criteria which determine how to link a pair also produces different results in different clustering results. For example, Complete linkage criteria chooses the maximum distance to link clusters, but Single linkage criteria chooses the minimum distance to link them. Thus, HCA results will vary by how to set up internal criteria. In addition, it doesn’t have to fix the number of clusters in advance, but its efficiency is relatively low. Deo et al. (2010) applied Hierarchical clustering method to metabolites profiling data about oral glucose tolerance test to cluster 50 samples. Hierarchical clustering has also used to classify the samples using GC-MS metabolic signature data of glioblastoma reflecting accelerated anabolic metabolism for a profiling study (Chinnaiyan et al., 2012).
k-means clustering: k-Means clustering assigns all samples to k different clusters where the number of the clusters, k, is known. k-Means clustering is also called ‘partitioning technique’. Suppose that n samples with p dimensional metabolite concentrations are partitioned to k different clusters of {C1,…,Ck} using k-Means clustering by minimizing within-cluster sum of squares as Eq. 10. This method begins by guessing initial means of k clusters and recursively updates clusters of each sample until there are no more changes in clusters of all samples:
| (10) |
where, μl are the means of clusters for l = 1,…,k. k-Means clustering algorithm is computationally efficient, but it has higher complexity order and the clustering performance is influenced by initial values. k-Means clustering technique results in different variants later such as fuzzy-k and PAM. Cuperlovic-Culf et al. (2009) applied fuzzy k-Means clustering method to group the type 2 diabetes patients with their NMR spectra of cancer cells for a fingerprinting study.
SOM: SOM (Self-Organizing Maps) which is also known as Kohonen neural network (Kohonen, 1982), can be also used for metabolomics data. Like k-Means clustering, SOM assumes the number of groups or clusters is known and it groups the data within this number k. SOM attempts to maintain the same topological relationship between the input space and the output space by projecting the vectors in the input space onto the output space where the input space is connected with the output space by nodes and the nodes have corresponding weights. Suppose that n input vectors whose lengths are p are mapped on the k vectors in the output space for typically k = n. This technique attempts to project the input space as similar as possible onto the output space by updating all weights on nodes based on ‘the winning node’ in tth learning process where the winning node is determined as the node whose distance is the minimum from a given input vector. The rule to update weights on each t can be summarized as follows (Mehrotra et al., 1996).
| • | Compute the distance between i input vector and a weight vector l which is associated with each output node for l = 1,…, k at t = t: |
| (11) |
Select the output node j* associated with the weight having the minimum distance.
Update weights to all nodes from j* using the following rule:
| (12) |
Make an increment of t.
Typically the learning rate η(t) is determined as 0<η(t)≤η(t-1)≤1. The applications of SOM with metabolomics data are relatively diverse from the correlation exploration to clustering (Steiner et al., 2002). SOM has applied to the correlation of GC-MS data to compare the morphology of 88 species of ants (Nikiforow et al., 2001) and H (Hierarchical)-SOM has used to classify 63 different nests of ants to 5 different species with GC-MS data (Nyamundanda et al., 2010). BL (Batch Learning)-SOM has also applied to a time-course metabolomics profiling with Arabidopsis thalinana cell culture for clustering the data matrix by salt--stress treatment using PCA to determine initial weights on nodes with GC-MS data (Kim et al., 2006).
CONCLUSION
Metabolomics is a relatively young but emerging science gaining attentions across all functional genomics. Current metabolomics studies primarily either accurately measure a small number of molecule to find key metabolites or analyze large numbers of compounds to identify the pathways or figure out the altered metabolism pattern from them. This paper begins with brief overviews about the current analytical methods and primary study approaches. Based on these overviews, the commonly applied bioinformatics methods to the metabolomics data are reviewed with their clinical examples.
Through the review of the methodological approaches, we can’t single one method out as the superior technique: Every method has its drawback and it can effect trivially in a certain situation, but it may affect significantly on the result on the contrary. However, there is little paper which observes the performance comparisons by different techniques in the same dataset. Because we can expect to lower False Discovery Rate (FDR) by finding the more suitable bioinformatics techniques, it is worth conducting the data-driven methods. Also, even though the data are generated by the same types of analytical platforms and the study approaches are the same, metabolites concentrations may be different patterns which may result in different performances by different techniques in the same category. In this case, more data-driven bioinformatics approaches may interpret the metabolomics data better. In addition, the current analytical techniques have much room to be improved. It is obvious that analytical techniques need to be improved which can satisfy both the coverage of metabolites and the accuracy of detection to facilitate further development in metabolomics studies. Yet, seeing on Table 1, the detection accuracy is not the best when the coverage is high and vice versa under the current technology. The analytical techniques could affect the quality of data, so they need to be chosen considering the study purpose. Besides analytical science, the improvement of the data interpretation is also important and bioinformatics does a key role. Especially, more advanced bioinformatics methods and their clinical applications are essential tools to exploit novel profiling techniques and to speed up the discoveries of unknown biomarkers. The bioinformatics techniques also may be applied to compensate for the weakness of the current analytical technologies. For example, peak shift or unidentified peaks may be solved by more diverse or mixed applications of bioinformatics techniques. Such as latent variable modeling using Factor analysis or mixture modeling may help to consider the hidden peaks in the interpretation.
Moreover, the use of more integrated information across other functional genomics fields may also help extracting more information from the metabolomics data. It is a still a burden handling high-dimensional data, but these approaches would help to speed up identifying the biomarkers for specific diseases and finding more vulnerable metabolic pathways. Thus, it is necessary to understand the bioinformatics techniques well and continue to seek for new solutions in metabolomics studies.
REFERENCES
- Allen, J., H.M. Davey, D. Broadhurst, J.K. Heald, J.J. Rowland, S.G. Oliver and D.B. Kell, 2003. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat. Biotechnol., 21: 692-696.
CrossRef - Altmaier, E., S.L. Ramsay, A. Graber, H.W. Mewes, K.M. Weinberger and K. Suhre, 2008. Bioinformatics analysis of targeted metabolomics: Uncovering old and new tales of diabetic mice under medication. Endocrinology, 149: 3478-3489.
CrossRefDirect Link - Barker, M. and W. Rayens, 2003. Partial least squares for discrimination. J. Chemometrics, 17: 166-173.
CrossRefDirect Link - Baumgartner, C., M. Osl, M. Netzer and D. Baumgartner, 2011. Bioinformatic-driven search for metabolic biomarkers in disease. J. Clin. Bioinform., Vol. 1.
CrossRefDirect Link - Blekherman, G., R. Laubenbacher, D.F. Cortes, P. Mendes and F.M. Torti et al., 2011. Bioinformatics tools for cancer metabolomics. Metabolomics, 7: 329-343.
CrossRef - Bogdanov, M., W. Matson, L. Wang, T. Matson, R. Saunders-Pullman, S.S. Bressman and M.F. Beal, 2008. Metabolomic profiling to develop blood biomarkers for Parkinson's disease. Brain, 131: 389-396.
CrossRef - Boros, L.G., D.J. Brackett and G.G. Harrigan, 2003. Metabolic biomarker and kinase drug target discovery in cancer using Stable Isotope-based Dynamic Metabolic Profiling (SIDMAP). Curr. Cancer Drug Targets, 3: 445-453.
CrossRef - Britz-McKibbin, P. and S. Terabe, 2002. High-sensitivity analyses of metabolites in biological samples by capillary electrophoresis using dynamic pH junction-sweeping. Chem. Rec., 2: 397-404.
CrossRef - Bylesjo, M., M. Rantalainen, O. Cloarec, J.K. Nicholson, E. Holmes and J. Trygg, 2006. OPLS discriminant analysis: Combining the strengths of PLS-DA and SIMCA classification J. Chemom., 20: 341-351.
CrossRefDirect Link - Cheng, L.L., C.L. Lean, A. Bogdanova, S.C. Wright Jr., J.L. Ackerman, T.J. Brady and L. Garrido, 1996. Enhanced resolution of proton NMR spectra of malignant lymph nodes using magic-angle spinning. Magn. Reson. Med., 36: 653-658.
CrossRef - Chinnaiyan, P., E. Kensicki, G. Bloom, A. Prabhu and B. Sarcar et al., 2012. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res., 72: 5878-5888.
CrossRef - Cuperlovic-Culf, M., N. Belacel, A.S. Culf, I.C. Chute and R.J. Ouellette et al., 2009. NMR metabolic analysis of samples using fuzzy K-means clustering. Magn. Reson. Chem., 47: S96-S104.
CrossRef - Deo, R.C., L. Hunter, G.D. Lewis G. Pare and R.S. Vasan et al., 2010. Interpreting metabolomic profiles using unbiased pathway models. PLoS Comput. Biol., Vol. 6.
CrossRefDirect Link - Fiehn, O., 2001. Combining genomics, metabolome analysis and biochemical modelling to understand metabolic networks. Comp. Funct. Genomics, 2: 155-168.
CrossRef - Fleming, C.M., B.R. Kowalski, A. Apffel and W.S. Hancock, 1999. Windowed mass selection method: A new data processing algorithm for liquid chromatography-mass spectrometry data. J. Chromatogr. A, 849: 71-85.
CrossRef - Goodacre, R., S. Vaidyanathan, W.B. Dunn, G.G. Harrigan and D.B. Kell, 2004. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol., 22: 245-252.
CrossRefDirect Link - Halket, J.M., A. Przyborowska, S.E. Stein, S.E. Stein, W.G. Mallard, S. Down and R.A. Chalmers, 1999. Deconvolution gas chromatography/mass spectrometry of urinary organic acids-potential for pattern recognition and automated identification of metabolic disorders. Rapid Commun. Mass Spectrom., 13: 279-284.
CrossRef - Johansen, K.K., L. Wang, J.O. Aasly, L.R. White and W.R. Matson et al., 2009. Metabolomic profiling in LRRK2-related Parkinson's disease. PLoS ONE, Vol. 4.
CrossRefDirect Link - Katajamaa, M., J. Miettinen and M. Oresic, 2006. MZmine: Toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics, 22: 634-636.
CrossRef - Katajamaa, M. and M. Oresic, 2007. Data processing for mass spectrometry-based metabolomics. J. Chromatography A, 1158: 318-328.
CrossRef - Kim, J.K., T. Bamba, K. Harada, E. Fukusaki and A. Kobayashi, 2006. Time-course metabolic profiling in Arabidopsis thaliana cell cultures after salt stress treatment. J. Exp. Bot., 58: 415-424.
CrossRef - Lucio, M., A. Fekete, C. Weigert, B. Wagele and X. Zhao et al., 2010. Insulin sensitivity is reflected by characteristic metabolic fingerprints: A fourier transform mass spectrometric non-targeted metabolomics approach. PLoS ONE, Vol. 5.
CrossRefDirect Link - Nyamundanda, G, L. Brennan and I.C. Gormley, 2010. Probabilistic principal component analysis for metabolomic data. BMC Bioinf., Vol. 11.
CrossRef - Qiu, Y., G. Cai, M. Su and T. Chen and Y. Liu et al., 2010. Urinary metabonomic study on colorectal cancer. J. Proteome Res., 9: 1627-1634.
CrossRef - Scalbert, A., L. Brennan, O. Fiehn, T. Hankemeier and B.S. Kristal et al., 2009. Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics, 5: 435-458.
CrossRef - Schripsema, J., 2010. Application of NMR in plant metabolomics: Techniques, problems and prospects. Phytochem. Anal., 21: 14-21.
CrossRef - Seierstad, T., K. Roe, B. Sitter, J. Halgunset and K. Flatmark et al., 2008. Principal component analysis for the comparison of metabolic profiles from human rectal cancer biopsies and colorectal xenografts using high-resolution magic angle spinning 1H magnetic resonance spectroscopy. Mol. Cancer, Vol. 7.
CrossRef - Vladimir, S., 2006. Metabolomics technology and bioinformatics. Brief. Bioinform., 7: 128-139.
CrossRef - Smith, C.A., J. Elizabeth, G. O'Maille, R. Abagyan and G. Siuzdak, 2006. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching and identification. Anal. Chem., 78: 779-787.
CrossRef - Spraul, M., P. Neidig, U. Klauck, P. Kessler and E. Holmes et al., 1994. Automated reduction of NMR spectroscopic data for statistical and pattern recognition classification of samples. J. Pharm. Biomed. Anal., 12: 1215-1225.
CrossRef - Steiner, F.M., B.C. Schlick-Steiner, A. Nikiforov, R. Kalb and R. Mistrik, 2002. Cuticular hydrocarbons of Tetramorium ants from central Europe: Analysis of GC-MS data with self-organizing maps (SOM) and Implications for systematics. J. Chem. Ecol., 28: 2569-2584.
CrossRef - Steuer, R., J. Kurths, O. Fiehn and W. Wechwerth, 2003. Observing and interpreting correlations in metabolomic networks. Bioinformatics, 19: 1019-1026.
CrossRef - Szymanska, E., E. Saccenti, A.K. Smilde and J.A. Westerhuis, 2012. Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics, 8: 3-16.
CrossRef - Trygg, J. and S. Wold, 2002. Orthogonal projections to latent structures (O-PLS). J. Chemometr., 16: 119-128.
CrossRef - Tsugawa, H., Y. Tsujimoto, M. Arita, T. Bamba and E. Fukusaki, 2011. GC/MS based metabolomics: development of a data mining system for metabolite identification by using soft independent modeling of class analogy (SIMCA). BMC Bioinform., Vol. 12.
CrossRef - Van Velzen, E.J.J., J.A. Westerhuis, J.P.M. van Duynhoven, F.A. van Dorsten and H.C.J. Hoefsloot et al., 2008. Multilevel data analysis of a crossover designed human nutritional intervention study. J. Proteome Res., 7: 4483-4491.
CrossRef - Viant, M.R., E.S. Rosenblum, R.S. Tieerdema, 2003. NMR-based metabolomics: A powerful approach for characterizing the effects of environmental stressors on organism health. Environ. Sci. Technol., 37: 4982-4989.
CrossRef - Vogels, J.T.W.E., A.C. Tas, J. Venekamp and J. van der Greef, 1996. Partial linear fit: A new NMR spectroscopy preprocessing tool for pattern recognition applications. J. Chemom., 10: 425-438.
CrossRef - Windig, W., J.M. Phalp and A.W. Payne, 1996. A noise and background reduction method for component detection in liquid chromatography/mass spectrometry. Anal. Chem., 68: 3602-3606.
CrossRef - Wold, S., M. Sjostrom and L. Erikson, 2001. PLS-regression: A basic tool of chemometrics. Chemometrics Intell. Lab. Syst., 58: 109-130.
CrossRefDirect Link - Wopereis, S., C.M. Rubingh, M.J. van Erk, E.R. Verheij and T. van Vliet et al., 2009. Metabolic profiling of the response to an oral glucose tolerance test detects subtle metabolic changes. PLoS, Vol. 4.
CrossRef - Yang, C., A.D. Richardson, J.W. Smith and A. Osterman, 2007. Comparative metabolomics of breast cancer. Pac. Symp. Biocomput., 12: 181-192.
PubMedDirect Link - Nikiforow, A., B. Schlick-Steiner, F. Steiner, R. Kalb and R. Mistrik, 2001. Classification of GC-MS data of epicuticular hydrocarbon from Tetramorium ants by self-organizing maps for morphological determinations. Proceedings of the 49th ASMS Conference on Mass Spectrometry and Allied Topics, May 27-31, 2001, Chicago, IL., USA.