
Abdullah A. Abba
Department of Medicine,
College of Medicine and King Khalid University Hospital: P.O Box 50726, Riyadh 11533, Kingdom of Saudi Arabia
Journal of Medical Sciences
Year: 2006 | Volume: 6 | Issue: 4 | Page No.: 704-708







ABSTRACT
Mitochondrial disease, a recently recognised and rapidly expanding field of medicine, has variable presentation. A spectrum of conditions has been described as purely a consequence of mitochondrial defect(s) while others are presumed to be causally related. We present a case of mitochondrial disease presenting with respiratory failure and discuss the respiratory manifestations of this condition and its management. There is a need for awareness of the characteristics of respiratory manifestations of the mitochondrial diseases.
PDF Abstract XML References
How to cite this article
Abdullah A. Abba, 2006. Mitochondrial Encephalomyopathy Presenting with Respiratory Failure in an Adult. Journal of Medical Sciences, 6: 704-708.
DOI: 10.3923/jms.2006.704.708
URL: https://scialert.net/abstract/?doi=jms.2006.704.708
DOI: 10.3923/jms.2006.704.708
URL: https://scialert.net/abstract/?doi=jms.2006.704.708
INTRODUCTION
Since the discovery of the genetic bases of mitochondrial diseases in 1988 (Holt et al., 1988; Wallace, 1988) there has been tremendous development in this subspecialty of medicine which is now referred to as mitochondrial medicine and the over 40 different identified diseases as mitochondrial cytopathies. Mitochondria are central to cellular function, being the part of the cell where oxygen and nutrients are converted to energy. Defects in any of the pathways may result in disease. Organs with greatest requirements for oxidative energy such as the heart, brain and muscles are therefore particularly vulnerable. Although most patients initially present to the neurologist, any organ system may be affected. Such patients may present to the pulmonary and critical care physician, hence the need for awareness of these conditions by these physicians. Respiratory failure, sleep apnoea, exercise intolerance/muscle weakness and lactic acidosis are some of the modes of presentation.
We describe a patient of mitochondrial myopathy who presented with acute respiratory failure and review the subject of mitochondrial encephalomyopathies.
CASE HISTORY
A 15-year old Saudi boy presented to the Emergency Room (ER) of Riyadh Medical Complex with a two-day history of cough and fever. Cough was productive of yellowish sputum and fever was continuous. He was seen a day earlier in the same facility with the same symptoms when he was given a course of antibiotics and some cough syrup without improvement.
The family gave history that patient was on medications for epilepsy which was described as myoclonic in nature since the age of eight years. He developed difficulties with walking ten years earlier and now is only able to crawl around the house. He became deaf at the age of 10 years and blind at the age of 13 years. The rest of systemic review was uneventful. A sister and a brother aged 18 and 12 years respectively have difficulties with walking but are largely independent.
Examination revealed patient to be gasping, cyanosed and drowsy. He was febrile at 38.2°C, tachypnoeic with respiratory rate of 34 min. His blood pressure was 160/100 mm Hg and pulse rate 140 min-1. Examination of the chest did not reveal any skeletal deformities. There were widespread crepititations in both axillary and infrascapular regions. Examination of the abdomen was unremarkable and the cardiovascular system was normal except for sinus tachycardia. Examination of the central nervous system revealed immature cataracts in both eyes.
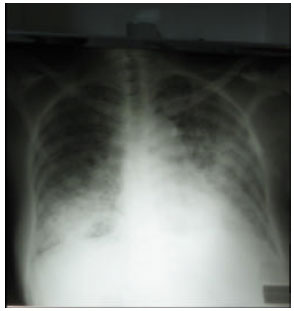 | |
| Fig. 1: | Chest radiograph showing extensive bronchopneumonia |
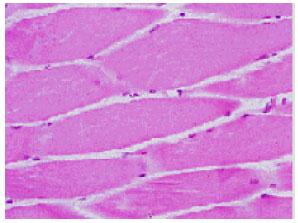 | |
| Fig. 2: | Muscle biopsy showing Red Ragged Fibers (RRF) |
Full evaluation of the motor, sensory and cerebellar system was not possible due to the critical state of the patient. There were callosities in both feet.
Investigations revealed leucocytosis with a white cell count of 15.6x103 L-1, haemoglobin of 10.9 g dL-1 and normal platelets. His biochemical profile was normal including the bone profile. His chest x-ray shows bilateral extensive airspace disease (Fig. 1) and his arterial blood gases were as follows: pO2 51 mm Hg, pCO2 69 mm Hg, pH 7.18, HCO3 20 mEq L-1 and SaO2 75%. The electrocardiogram revealed sinus tachycardia and right bundle branch block. Patient shortly after presentation developed cardio respiratory arrest and was intubated and ventilated.
He was admitted with a diagnosis of community-acquired pneumonia with respiratory failure. His management included ventilatory support and antibiotics (ceftriaxone, erythromycin and clindamycin). He made remarkable improvement and was afebrile on the second day and chest examination was clear on the seventh day. A repeat chest x-ray was normal. He, however, could not be weaned from the ventilator because of hypercapnia and hypoxia. With the history of myclonic epilepsy, deafness, blindness and a positive family history, mitochondrial myopathy was suspected. A muscle biopsy was done which revealed the characteristic red ragged fibres (Fig. 2). Lactic acid levels were also elevated at 3.8 mmol L-1 (normal range 0.67-1.8 mmol L-1).
Patient remained on ventilator as all efforts to wean him failed. He subsequently developed malignant arrhythmias and could not be revived on the 120th day of admission.
DISCUSSION
Primary mitochondrial diseases (MD) remain uncommon and the prevalence is believed to be 10 to 15 cases per 100,000 persons (Chinnery and Turnbull, 2001). Mitochondrial involvement is however being investigated in a wide spectrum of diseases including Alzheimer’s dementia and Parkinson’s disease. Normal cellular function is dependant on adequate amounts of Adenosine Triphosphate (ATP). Lack of ATP not only leads to cellular malfunction but also to alternative pathways of metabolism. This in turn leads to accumulation of toxic products such as lactate. MD therefore affects mainly the highly metabolically active cells such as the heart, brain and skeletal muscles. There is a broad spectrum of expression of disease ranging from the mild to severe. Similarly any age group can be affected.
The genetics of MD is different from mendelian genetics in three respects:
| • | Inheritance is maternal-because all mitochondria and mitochondrial DNA in the zygote derive from the ovum. A mother carrying mtDNA mutation passes it to all her children but only the daughters will transmit to their progeny. Recently however, there has been a report of paternal transmission (Schwartz and Vissing, 2002) |
| • | Heteroplasmy-a situation in which one mitochondrion harbours both normal and mutant mDNA. Cellular dysfunction occurs and clinical signs become apparent only when a minimal number of mutant mtDNA exist in a cell. This is the threshold effect and is lower in highly metabolically active tissues. |
| • | Mitotic segregation: during mitosis, the proportion of mutant mtDNA received by daughter cells may vary. If the threshold is exceeded disease may manifest. This explains the age-related and tissue-related variability of clinical features. |
In MD, therefore any mode of inheritance can be observed: sporadic, autosomal dominant or recessive or maternal inheritance. Similarly, any age group may be affected and the severity of disease may vary widely. No organ system is sacrosanct.
Respiratory failure as in our patient may be the initial sign of the disease. It may be fulminant or intermittent and relapsing. Two presentations are recognised, first, muscle fatigue following a period of increasing dyspnoea which may masquerade as exacerbation of COPD. Secondly, it may present as hypoventilation as result of an inciting event such pneumonia, low doses of sedatives-hypnotics or high altitude. The later seems to be the case in our patient. The mechanism of hypoventilation may be multifactorial including impaired response to hypoxia and hypercapnia as demonstrated in a study by Carroll et al. (1976), muscle weakness (Cros, 1992) and diaphragmatic paralysis (Desmuelle, 1988).
The prevalence of respiratory failure is high therefore this mode of presentation of MD may be missed. The index of suspicion should however be high in the following circumstances: failure to wean from ventilator when other causes such as myasthenia gravis, hypothyroidism, electrolyte disturbances and critical illness polyneuropathy are ruled out; respiratory failure following minimal sedation; or persistent lactic acidosis in the absence of hypoperfusion or drugs known to cause lactic acidosis (Clay, 2001).
There is variable response to mechanical ventilation. Some patients recover with intermittent relapses, others are partially dependant on ventilator while others remain permanently dependant. Our patient fell in the last category and died from cardiac arythymias. Various degrees of heart block have been described and because of the frequency of fatal conduction defects, placement of prophylactic pacemaker is recommended (Polak, 1989).
Diagnosis of MD is reached after consideration of the clinical features including a complete history (with emphasis on family history) and physical examination. Laboratory tests are used to determine disruption of normal oxidative phosphorylation. A lactate-to-pyruvate ratio of more than 20 is abnormal. Twenty-four-hour measurement of pyruvate, lactate, glucose, phosphate and amino acids may detect defects in the renal tubular cells which are highly dependent on oxidative phosphorylation (Shoffner, 1999).
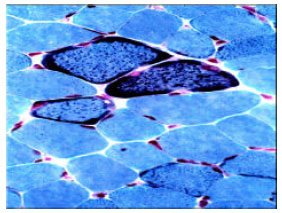 | |
| Fig. 3: | Muscle biopsy showing RRF (Gomori trichrome; original x 500). Source: Clay et al. (2001) |
Non-invasive tests that may be helpful in diagnosis include pulmonary function tests which may show reduced forced vital capacity, maximum minute ventilation and inspiratory/expiratory pressures due to muscle weakness. Exercise testing may demonstrate elevated heart rate relative to degree of work and a diminished maximal workload; reduced maximal oxygen consumption (VO2max) and ventilatory threshold and increased respiratory exchange ratio. These changes are non-specific. 31P nuclear magnetic resonance spectroscopy is used to assess the metabolic state of muscle fibres. In MD, the resting phospocreatine (PCr) level is reduced and rapidly declines during exercise and the post exercise recovery of PCr, adenosine diphosphate(ADP) and inorganic phosphate (Pi) is prolonged.
Confirmation of the diagnosis is from muscle biopsy specimens which traditionally show red ragged fibres and sometimes lipid deposition on light microscopy (Fig. 2). Staining with Gomori’s trichrome makes the appearance more striking (Fig. 3). These fibers may however be absent in missense mutations, nuclear mutations,and CPT ddeficiency. On the other hand, they are not specific to MD and may occur in small quantities in Duchenne’s muscular atrophy, neurogenic dystrophy, polymyositis and dermatomyositis (Clay, 2001). Electron microscopy may show abnormal mitochondria with abnormal cristae and /or paracrystalline inclusions. Mitochondrial enzymology can also be done on muscle biopsy specimens to locate specific abnormalities.
If suspicion remains high and the laboratory, non-invasive and biopsy muscle specimens are normal, then genetic analysis of mtDNA is warranted. This may involve screening the genome by in situ hybridization for more common mtDNA mutations or the entire genome may be sequenced.
Current treatment strategies are largely supportive and MD usually progress relentlessly to disability and death. A recent meta-analysis of treatment trials concluded that there is currently no clear evidence supporting the use of any intervention in mitochondrial disorders (Chinnery; 2006). Measures that have been tried include avoidance of precipitating factors such as fasting, exposure to cold, alcohol, tobacco, sedatives, anaesthesia infections, over exertion and stress. High carbohydrate diet shifts metabolism towards glycolysis and this is one aspect that may be recommended to the patients. Regulated aerobic exercises limited to 60-80% capacity may have a direct effect on the population of mtDNA (Taivassalo, 1998). Drugs, vitamins and food supplements aimed at improving ATP production by providing protein for oxidative phosphorylation and inhibiting lactate production have been tried with minimal effects. These include riboflavin, co-enzyme Q10 or ubiquinone, dichloroacetate and dimethyl glycine. Further research is needed to establish the role of a wide range of therapeutic options including both general measures and drugs.
ACKNOWLEDGMENTS
We wish to thank Chest, The Official Journal of American College of Chest Physicians for their kind permission to reproduce Fig. 3 which appeared in the article referenced (Clay, 2001).
REFERENCES
- Schwartz, M. and J. Vissing, 2002. Paternal inheritance of mitochondrial DNA. New Engl. J. Med., 347: 576-580.
PubMed