
Hala T. El-Bassyouni
Department of Clinical Genetics, National Research Centre, Cairo, Egypt
Adel M. Ashour
Department of Clinical Genetics, National Research Centre, Cairo, Egypt
Afaf Ezzat
Department of Food Science Nutrition, National Research Centre, Cairo, Egypt
Randa Bassiouni
Department of Clinical Genetics, National Research Centre, Cairo, Egypt
Ekram M. Fateen
Department of Biochemical Genetics, National Research Centre, Cairo, Egypt
Journal of Medical Sciences
Year: 2006 | Volume: 6 | Issue: 3 | Page No.: 452-457







ABSTRACT
Galactosemia is an autosomal recessively inherited disorder of galactose metabolism. It has good prognosis, if detected in neonatal period or early infancy. Treatment consists of life long dietary restriction of galactose. Present study included eight patients with galactosemia on dietary treatment, five of them had galactose-1-phosphate uridyltransferase deficiency known as classical galactosemia and three had uridine-diphosphate galactose-4` epimerase deficiency. Clinical evaluation of patients under galactose restricted diet and assessment of the antioxidant status in response to dietary therapy was done. Delayed milestones were present in all patients, jaundice at birth was present in 4 and low birth weight was present in 3 patients. Craniofacial dysmorphism was present in 5 patients. Hepatomegaly was present in 6 patients. MRI of the brain showed brain atrophy in 3 patients and demyelination in 2 patients. There was cataract in 7 patients. The levels of zinc, copper, iron, calcium, phosphate, magnesium, selenium, manganese, ß-carotene and vitamin A were evaluated in the blood of galactosemic patients on galactose restricted diet and a comparison between trace elements, ß-carotene and vitamin A in studied patients with galactosemia and controls was done. Copper, calcium, phosphate, manganese and ß-carotene levels in blood were significantly decreased in our patients (p<0.001) than in controls. These findings suggest that patients on galactose restricted diet are at risk of oxidative stress. The data emphasize the importance of dietary supplementation with an antioxidant containing ß-carotene, calcium, copper, selenium and manganese to inhibit oxidative stress in these patients. Consequently this will minimize the neurological deficits improve bone mineralization, reduce the development of retinopathy and damage to liver cells in patients with galactosemia.
PDF Abstract XML References Citation
How to cite this article
Hala T. El-Bassyouni, Adel M. Ashour, Afaf Ezzat, Randa Bassiouni and Ekram M. Fateen, 2006. The Effect of Diet on Antioxidant Status in Patients with Galactosemia. Journal of Medical Sciences, 6: 452-457.
DOI: 10.3923/jms.2006.452.457
URL: https://scialert.net/abstract/?doi=jms.2006.452.457
DOI: 10.3923/jms.2006.452.457
URL: https://scialert.net/abstract/?doi=jms.2006.452.457
INTRODUCTION
Galactosemia is an autosomal recessive inherited disorder of galactose metabolism, which occurs as a consequence of a deficiency of one of three principal enzymes involved in the metabolism of galactose, through its conversion to glucose. These enzymes are galactose-1-phosphate uridyltransferase (GALT), uridine-diphosphate galactose-4' epimerase and galactokinase (Wasilenko et al., 2005). The most common deficiency in all communities is that of the transferase enzyme and it is this enzyme deficiency that underlies "classical galactosemia" (OMIM, 2004; Bosch et al., 2002).
Because milk is the staple of an infant's diet, early diagnosis and treatment of this disorder is absolutely essential to avoid serious lifelong disability. Despite the treatment, long term complications occur such as disturbed mental and motor development, decreased bone mineral density and learning disorders (Arn, 2003).
Failure to thrive is the most common initial clinical symptom of galactosemia. Vomiting or diarrhoea usually begins within a few days of milk ingestion. Jaundice of intrinsic liver disease may be accentuated by the severe haemolysis occurring in some patients (Huidekoper et al., 2005). The pathophysiology of cataracts is one of the well characterized aspects of galactosemia. Cataracts have been observed within a few days of birth. These may be found only on slit-lamp examination and missed with an ophthalmoscope, since they consist of punctate lesions in the fetal lens nucleus (Holton et al., 2001). The most vexing problem facing patients with galactosemia is the effect of galactose deficiency on brain development and function. The patient may have decreased IQ and/or learning disability. Clinically evident speech delay and cerebellar signs are more frequent than other findings. Abnormal white matter signal is found in most subjects, but this abnormality does not correlate with cognitive outcome. Webb et al. (2003) noted that verbal dyspraxia (chaotic speech) is found in many galactosemic patients.
Oxidative stress may be implied in the pathogenic mechanisms of galactosemia. The lower activities observed in patients on natural protein restriction may likely be due to a deficient bioavailability of antioxidant cofactors (Vilaseca-Busca et al., 2002). Dietary supplementation with an antioxidant containing β-carotene and selenium inhibits oxidative stress in galactosemia. Antioxidants inhibit the abnormal metabolic process that contributes to the development of the disease (Kowluru et al., 2000).
Iron homeostasis and galactose metabolism are linked with one another because galactose metabolism is defective when iron availability is restricted. Iron deficient galactosemic patients might be more severely compromised than iron repleted individuals (Shi et al., 2003).
Treated galactosemic children are at risk of abnormal bone mineralization which may predispose them to osteoporosis and fracture in adult life (Rubio-Gozalbo et al., 2002). Also, calcium is important in relation to maintenance of lens transparency (Kamei et al., 1987).
Nutrition plays a vital role in maintaining physical growth and development throughout life. The dietary modifications form the mainstay to disease stabilization and control. A modified diet is essential for the patient's survival and adequate mental development (Badawy et al., 2003). In the last decade there has been tremendous progress in cloning of various genes, identification of their protein products and detection of different mutations in many metabolic disorders. With improved understanding of the underlying metabolic pathways, dietary interventions may be more appropriately targeted (Bavdekar et al., 2002).
MATERIALS AND METHODS
The study included eight galactosemic patients on galactose restricted diet (seven males and one female). They were recruited between 2003 and 2005 from the Clinical Genetics Department, National Research Centre, Cairo, Egypt. All the patients have been diagnosed in the neonatal period and had good dietary control. Their age of onset ranged from 10 days to 30 days.
The control group consisted of ten healthy controls who were matching for age and sex.
Each patient was subjected to meticulous clinical examination to detect any malformation or anomaly. Three generations pedigrees were constructed and analysed with special emphasis on positive parental consanguinity and similarly affected sibs in the family.
In addition the following investigations were done to all the patients:
| • | Abdominal sonography. |
| • | MRI brain. |
| • | Complete eye evaluation. |
| • | Cytogenetic analysis of peripheral blood lymphocytes from the patients was done using G-banding technique. |
| • | Biochemical assessment of: Trace elements (Zn, Cu, Fe, Ca, P, Mg, Se and Mn) and antioxidants (β-carotene and vitamin A). The levels of plasma zinc and copper were measured according to the methods of Homsher and Zak (1985) and Abe et al. (1989), respectively. The determination of iron was according to the method of Tabacco et al. (1981). Calcium and phosphorus were measured according to the method of Gitelman (1967) and Gamst and Try (1980), respectively. The level of magnesium was measured according to the methods of Gindler and Heath (20). The level of selenium and manganese were measured with a Varian Spectr AA 220 atomic absorption spectrometer equipped with a graphite-surnace tube atomizer (GTA). Samples and standard were injected into GTA using an auto sampler (GTA 100). The levels of β-carotene and vitamin A were determined according to the method of John et al. (1963). |
| • | Determination of total galactose (galactose and galactose 1-phosphate) in dried blood samples according to the method of Diepenbrock et al. (1992). The enzyme levels of galactose-1-phosphate uridyltransferase and uridine diphosphate galactose-4-epimerase were estimated according to the method of Shin (1991). |
RESULTS
This study was carried out on eight galactosemic patients on restricted galactose dietary treatment (seven were males and one female). Positive consanguinity was present in 5 patients (62.5%), there was no similarly affected members in the families.
Table 1 shows the clinical findings in the studied patients. There were five patients with Galactose 1-uridyl Transferase Deficiency (GALT) and the other three with epimerase deficiency. The mode of inheritance in both groups is autosomal recessive. Among the groups, seven were males and one female with parental consanguinity in five patients (62.5%). Delayed milestones were present in all patients, jaundice at birth was present in 4 (50%) and low birth weight was present in 3 patients (37.5%). Craniofacial dysmorphism was present in 5 patients (62.5%) in the form of frontal bossing, depressed nasal bridge and low set ears. In patient (1), there was incomplete simian crease in left hand and bilateral talipes equinovarus. In patient (4) there was arachnodactyly of the fingers and dentinogenesis imperfecta. In patient (6) there was bilateral simian crease, rocker bottom feet and left inguinal hernia. In patient (7) there was bilateral clinodactyly of fourth finger. However, chromosomal analysis of all patients was normal.
Hepatomegaly was present in 6 patients (75%), patient (8) showed paucity of the interlobular ducts in his liver biopsy.
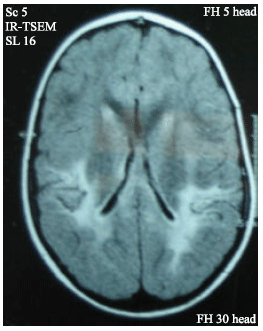 | |
| Fig. 1: | MRI T2 axial section showing white matter demyelination around the occipital horn of lateral ventricle in patient number 1 |
He had no cardiovascular, vertebral or renal abnormalities which excludes Alagille syndrome. MRI of the brain showed brain atrophy in 3 patients (3, 5 and 6), demyelination in 2 patients (1 and 8) and degeneration of basal ganglia in one patient (7). There was cataract in 7 patients (87.5%) three of them had squint on top (1, 3 and 5).The ERG in patient (8) showed diffuse retinal pathology affecting rods and cones.
The Table 2 shows significant decrease in the level of Cu, Ca, P, selenium, Mn and β-carotene in patients compared to the control group.
MRI T2 axial section showing white matter demyelination around the occipital horn of lateral ventricle Fig. 1.
DISCUSSION
Galactosemia is an autosomal recessive disorder of galactose metabolism caused by a deficiency of one of three principal enzymes involved in the metabolism of galactose through its conversion to glucose. These enzymes are galactose-1-phosphate uridyltransferase (GALT), uridine-diphosphate galactose-4' epimerase and galactokinase. In our study, galactose-1-phosphate uridyltransferase enzyme was deficient in five patients while epimerase deficiency was present in three. Because galactosemia is a relatively rare disease with an average of 3 new cases per year in our clinic, the patients group participating in this study was small.
| Table 1: | Clinical picture and enzyme deficiency |
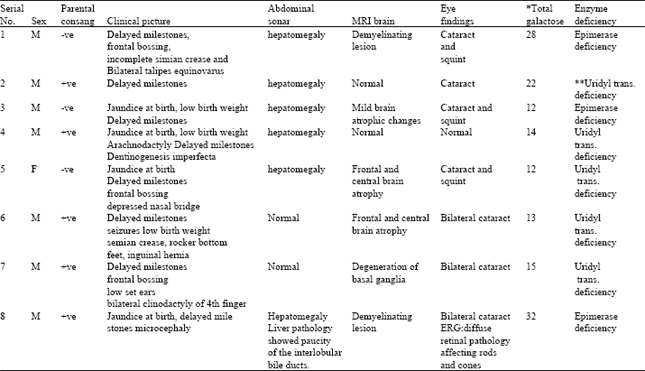 | |
| * Total galactose in blood (normal value: 0-5 mg dL-1) **Uridyl transferase deficiency | |
| Table 2: | Comparison between trace elements, β-carotene and vitamin A in studied patients with galactosemia and controls |
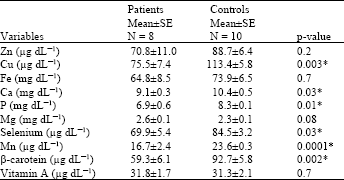 | |
| *p-value is significant if ≤0.05 | |
Parental consanguinity was present in five patients (62.5%). Several families with epimerase deficiency have been reported, but most are from highly consanguineous families and there does not appear to be a consistent phenotype which can clearly point to epimerase deficiency (Schulz et al., 2005).
Long-term results of treatment have been disappointing; IQ is low in many despite early and seemingly adequate therapy (Bosch et al., 2004). For example, the retrospective study by Schweitzer et al. (1993) reports the cause of the unsatisfactory outcome of seemingly good control of galactose intake and the disturbances in long-term development despite treatment was unclear. Possibilities include chronic intoxication by galactose metabolites or deficiency of galactose-containing glycoproteins and/or glycolipids as a result of an over restrictive galactose-free diet. However, Badawy et al. (2003) found that 80% of patients had improved both in mental and motor functions with galactose free diet.
Increased oxidative stress may be involved in the complications of galactosemia (Schulpis et al., 2005). Antioxidant supplementation can help alleviate the subnormal activities of antioxidant defense enzymes (Renu et al., 1999).
The results of assessment of the antioxidant minerals (Zn, Cu, Fe, Ca, P, Mg, Se and Mn ) and antioxidants (β-carotene and vitamin A) showed significant decrease in Cu, Ca, P, Se, Mn and β-carotene.
Kowluru et al. (2001) mentioned that β-carotene and selenium prevent galactosemia-induced elevation of retinal oxidative stress. Therefore, supplementation with antioxidants such as alpha-tocopherol, N-acetyl cysteine, ascorbic acid, beta-carotene and selenium can offer an achievable and inexpensive adjunct therapy to help inhibit the development of retinopathy as they are neuroprotective.
In this study, 6 patients presented with hepatomegaly (75%). Surgical biopsy of the liver showed marked fatty infiltration, periportal fibrosis and cirrhosis. Pozzato et al. (2005) studied patients with galactosemia who had enlarged liver but the patients did not show increased liver echogenicity. Marcoux et al. (2005) mentioned that galactosaemia reveals usually in the second and third weeks of life with a severe liver dysfunction. They reported a case of congenital galactosaemia with an early onset liver failure. Therefore, evaluation of patients with galactosaemia should not be limited to enzymatic analysis, but should also include hepatic imaging; especially ultrasonography (Nishimura et al., 2004). Patient number 8 showed paucity of the interlobular bile ducts in his liver biopsy. This coincides with the findings of Balistrari (1987) who mentioned that some syndromes characterised morphologically by intrahepatic cholestasis may be associated with paucity of the interlobular bile ducts. Schildkraut (2003) mentioned that the association of certain metabolic diseases with paucity of interlobular bile ducts carries a grave prognosis. Combined therapy with Coenzyme Q 10, Zn, Mg and selenium were effective in reducing damage to liver cells (Maritim et al., 2003).
MRI of the brain showed brain atrophy, demyelination and degeneration of basal ganglia. Similar findings were mentioned by Mizoguchi et al. (2001) who described manganese elevation in blood and magnetic response imaging changes in the basal ganglia of his patients.
These findings suggest that treated galactosemic patients are at risk of oxidative stress and abnormal bone mineralization. Therefore, therapeutic intervention in these cases should be more appropriately targeted. Present data emphasise the importance of antioxidants and trace elements to minimize the neurological deficits and improve bone mineralization, inhibit the development of retinopathy and reduce the damage to liver cells in patients with galactosaemia.
REFERENCES
- Abe, A., S. Yamashita and A. Noma, 1989. Sensitive, direct colorimetric assay for copper in serum. Clin. Chem., 35: 552-554.
PubMedDirect Link - Bavdekar, A., S. Bhave and A. Pandit, 2002. Nutrition management in chronic liver disease. Indian J. Pediatr., 69: 427-431.
CrossRefDirect Link - Bosch, A.M., H.D. Bakker, A.H.V. Gennip, J.V.V. Kempen, R.J.A. Wanders and F.A. Wijburg , 2002. Clinical features of galactokinase deficiency a review of literature. J. Inherited Metabol. Dis., 25: 629-634.
Direct Link - Bosch, A.M., A.G. Martha, H.D. Bakker, S.A. Heijmans, F.A. Wijburg and B.F. Last, 2004. Living with classical galactosemia health related quality of life consequences. Pediatrics, 113: 423-428.
PubMed - Diepenbrock, F., R. Heckler, H. Schickling, T. Engelhard, D. Bock and J. Sander, 1992. Colorimetric determination of galactose and galactose 1phosphate from dried blood. Clin. Biochem., 25: 37-39.
PubMed - Gamst, O. and K. Try, 1980. Determination of serum phosphorus without deproteinization by ultraviolet spectrophotometry of the phosphomolybdic acid complex. Scand. J. Clin. Lab. Invest., 40: 483-486.
Direct Link - Gitelman, H.J., 1967. An improved automatic procedure for the determination of calcium in biological specimens. Anal. Biochem., 18: 521-531.
CrossRef - Neeld, Jr. J.B. and W.N. Pearson, 1963. Macro- and micromethods for the determination of serum vitamin A using trifluoroacetic acid. J. Nutr., 79: 454-462.
PubMedDirect Link - Kowluru, R.A., J. Tang and T.S. Kern, 2001. Abnormalities of retinal metabolism in diabetes and experimental galactosemia effect of long term administration of antioxidants on the development of retinopathy. Diabetes, 50: 1938-1942.
Direct Link - Nishimura, Y., G. Tajima, A.D. Bahagia, A. Sakamoto and T. Saheki et al., 2004. Differential diagnosis of neonatal mild hypergalactosaemia detected by mass screening clinical significance of portal vein imaging. J. Inherit. Metab. Dis., 27: 11-18.
Direct Link - Pozzato, C., A. Curti, G. Radaelli, L. Fiori, S. Rossi, E. Riva and G. Cornalba, 2005. Abdominal ultrasonography in inherited diseases of carbohydrate metabolism. Radiol. Med. (Torino), 109: 139-147.
Direct Link - Renu, A. R. Kowluru, L. Ronald, Engerman, S. Timothy and S. Kern, 1999. Abnormalities of retinal metabolism in diabetes or experimental galactosemia VI Comparison of retinal and cerebral cortex metabolism and effects of antioxidant therapy. Free Radic. Biol. Med., 26: 371-378.
CrossRef - Schildkraut, V.Y., 2003. Nonsyndromic paucity of interlobular bile ducts report of 10 patients. J. Pediatr. Gastroenterol. Nutr., 37: 546-549.
Direct Link - Schulz, J.M., K.L. Ross, K. Malmstrom, M. Krieger and J.L.F. Keil, 2005. Mediators of galactose sensitivity in UDP galactose 4 epimerase impaired mammalian cells. J. Biol. Chem., 280: 13493-13502.
CrossRef - Schweitzer, S., Y. Shin, C. Jakobs and J. Brodehl, 1993. Long term outcome in 134 patients with galactosaemia. Eur. J. Pediat., 152: 36-43.
CrossRef - Shi, X., K. Chabarek, A. Budai and Z. Zhu, 2003. Iron requirement for GAL gene induction in the yeast saccharomyces cerevisiae. J. Biol. Chem., 278: 43110-43113.
Direct Link - Busca, M.A.V., R.A. Iriberri, C.C. Mallolas, N.B. Tarrau, J. Campistol, M.P. Marfa and C.S. March, 2002. Abnormal antioxidant system in inborn errors of intermediary metabolism. Rev. Neurol., 34: 1021-1024.
Direct Link - Wasilenko, J., M.E. Lucas, B. James, J.B. Thoden, H.M. Holden and J.L.F. Kei, 2005. Functional characterization of the K257R and G319E hGALE alleles found in patients with ostensibly peripheral epimerase deficiency galactosemia. Mol. Genet. Metabol., 84: 32-38.
CrossRef - Webb, A.L., R.H. Singh, M.J. Kennedy and L.J. Elsas, 2003. Verbal dyspraxia and galactosemia. Pediatr. Res., 53: 396-402.
Direct Link