
C.N. Fokunang
Structural Biology Unit School of Plant Sciences
University of Reading
P.O. Box 221 , Whiteknights
Reading, Berkshire, RG6 6AS
United Kingdom
K.A. Watson
Structural Biology Unit School of Plant Sciences
University of Reading
P.O. Box 221 , Whiteknights
Reading, Berkshire, RG6 6AS
United Kingdom
L. Skipper
Structural Biology Unit School of Plant Sciences
University of Reading
P.O. Box 221 , Whiteknights
Reading, Berkshire, RG6 6AS
United Kingdom
A. Purvis
Structural Biology Unit School of Plant Sciences
University of Reading
P.O. Box 221 , Whiteknights
Reading, Berkshire, RG6 6AS
United Kingdom
C. Browning
Structural Biology Unit School of Plant Sciences
University of Reading
P.O. Box 221 , Whiteknights
Reading, Berkshire, RG6 6AS
United Kingdom
Journal of Medical Sciences
Year: 2005 | Volume: 5 | Issue: 3 | Page No.: 141-152







ABSTRACT
The aim of this study was to amplify and clone the Nucleotide Binding Domain (NBD1) of Sulphonylurea Receptor 1 (SUR1) from hamster. SURI is a component of the ATP-sensitive K-channel (KATP channels) which play a central role in the control of insulin secretion from the β-pancreatic cells. The KOD HiFi DNA polymerase was used to amplify the gene encoding the NBD1 region of SUR1. This was then digested with restriction enzyme EcoRI and cloned into pETBlue-1 Blunt vector and subsequently into a larger pPIC9 (8023 base pair) vector. The cloned insert was transformed using NovaBlue Competent Cells. The analysis of transformants by double restriction digest using restriction enzymes SnaBI and XbaI confirmed that cloning was successful. The correct orientation of cloned NBD1 inserts were selected for the recombinant protein expression and purification into yeast Pichia pastoris and subsequently for X-ray crystallographic studies.
PDF Abstract XML References Citation
How to cite this article
C.N. Fokunang, K.A. Watson, L. Skipper, A. Purvis and C. Browning, 2005. Molecular Cloning of the Nucleotide Binding Domain of Sulphonylurea Receptor 1, a Component of the ATP-sensitive K-channel. Journal of Medical Sciences, 5: 141-152.
DOI: 10.3923/jms.2005.141.152
URL: https://scialert.net/abstract/?doi=jms.2005.141.152
DOI: 10.3923/jms.2005.141.152
URL: https://scialert.net/abstract/?doi=jms.2005.141.152
INTRODUCTION
ATP-sensitive K-channels (KATP channels) play a central role in the control of insulin secretion from pancreatic ß-cells[1-3]. Their closure in response to changes in intracellular adenine nucleotide concentrations couples changes in ß-cell glucose metabolism to membrane depolarisation and hence Ca2+-influx and insulin release. They are also closed by sulphonylureas such as tolbutamide, which are used in the treatment of Non-insulin-dependent Diabetes Mellitus (NIDDM)[1,4,5] and they are opened by diazoxide, a sulfonamide drug used to treat excessive insulin secretion found in insulinoma or Persistent Hypoglycaemia and Hyper-insulinaemia of Infants (PHHI)[5-7].
KATP-channels are made up of two components, a channel-forming subunit, Kir6.2 and a larger regulatory subunit, Sulfonylurea Receptor 1 (SUR1), which mediates the effect of sulphonylureas and diazoxide on channel activity[8,9]. SUR1, belongs to the ATP-binding Cassette (ABC) family of proteins and contains two putative nucleotide-binding folds which mediate activation of the channel by MgADP[10,11]. Current evidence suggests, however, that the major inhibitory effect of ATP is mediated by Kir6.2[12,5]. Studies of the KATP channels indicate that the active channel is an octomer consisting of four molecules each of Kir6.2 and SUR1[9,13]. Transient transfection of mammalian cells with SUR1 leads to the expression of high-affinity sulfonylurea binding sites. However, both subunits are required for the expression of active KATP channels in Xenopus oocytes or mammalian cells[7,10,14].
Nucleotide binding domains and hydrolysis: The location of the site to which ATP binds to cause KATP channel closure has been hotly debated. SUR1 contains two nucleotide binding domains with signature motifs for ATP recognition, whereas Kir6.2 has none[8,9,15]. The binding site for ATP does not lie solely on SUR1, but could also lie on Kir6.2, or possibly on a separate protein that interacts with the ATP channel and is endogenously expressed in the heterologous system used for expression of cloned channels[5,8,9].
Nucleoside diphosphates such as ADP and GDP have effects on KATP channel activity inhibition and activation. The latter, but not the former, requires the presence of Mg2+. Whereas the inhibitory effects of nucleotides probably involve interaction with Kir6.2, the stimulatory effects are mediated by the Nucleotide Binding Domains (NBDs) of SUR1. The NBDs of other ABC transporters have been shown to bind and hydrolyse ATP[16-18]. Each NBD contains three motifs that are important in this process: a Walker A (WA) motif, Walker B (WB) motif and a linker region[19,20]. A conserved, positively-charged lysine residue in the WA motif is involve in co-ordinating the negative charge of the phosphate tail of ATP, a negatively- charged aspartate residue in WB motif is believed to co-ordinate the Mg2+ ion of MgATP and the linker sequence might influence ATP-binding or hydrolysis and the mechanism by which conformational changes in the NBDs are transduced to other regions of the protein[9,21]. Electrophysiological studies have shown that mutating the WA lysine or WB aspartate in either of the NBDs of SUR1 has two consequences: MgADP-induced channel activation is abolished and sensitivity of the channel to ATP-mediated inhibition is slightly enhanced[11,14,22].
The aim of this study was to amplify and clone the Nucleotide Binding Domain (NBD1) of Sulphonylurea Receptor 1 (SUR1) from hamster. SURI is a component of the ATP-sensitive K-channel (KATP channels) which plays a central role in the control of insulin secretion from the β-pancreatic cells. Cloning of the NBD1 of SUR1 was important to optimize the expression and purification of the recombinant protein leading to X-ray crystallographic studies
MATERIALS AND METHODS
KOD HiFi Polymerase Chain Reaction (PCR)
Primer design: Three primers in total used for the amplification of SUR1 Nucleotide Binding Domain (NBD1) were designed. Primer was designed using the Primer 3 program on website: www.justbio.com. The forward primer, of 24-mer Tm 69.5°C, complementary to base 2003-2814 of the hamster SUR1 DNA, with a sequence that codes an EcoR1 restriction site. A reversed primer consisted of a poly linker between non coding and 6 histidine, with 72-mer, Tm >75°C. The second reversed primer was designed with a histidine, without poly linker, length 51-mer Tm >75°C. His-tag was needed in the reverse primers to enhance the protein purification process since pPIC-9 vector only enhance ligation process but plays no role in purification. The poly linker may be required to allow the 6x histidine to remain outside protein molecule. The stop codon was required to stop the polymerase reaction and EcoRI restriction enzyme was needed because they do not cut in coding region. Two extra base pairs were required on all primers at the 5' position for restriction enzyme to bind. To ensure accurate replication, a polymerase with a highly progressive 3' to 5' exonuclease activity was required. The KOD HiFi DNA Polymerase (Novagen) was the enzyme of choice providing satisfactory amplification.
The PCR reaction product was gently mixed and centrifuge briefly to bring the reaction components to the bottom of the tube. The tubes were capped and placed in thermal cycler.
PCR reaction condition: Denature 20 sec at 98°C; Anneal-30 sec at 68°C for NBD1 with his-tag reverse primer and 65°C for his-tag plus poly-linker reverse primer. Extention-None; Number of cycles 30.
To analyze the reaction products, a 20 μL sample was removed and added to 5 μL of loading buffer.
Agarose gel electrophoresis: Six gram agarose was dissolved in 500 mL tris acetate (1xTAE buffer), made earlier from 10 mL 50x TAE solution in 490 mL water. This was microwaved to melt agarose solution and then transferred to a water bath under temperature condition of 55°C, for 20 min. For a small agarose gel, 40 mL of agarose solution was mixed with 1 μL ethidium bromide and poured in well, then allowed to set. Solidified gel was transferred to an electrophoretic tank and 300 mL of 1 x TAE buffer poured to fill the gel tank. Loading gel was done with a 10 μL ladder, 25 μL PCR products and ran at 90 volt, 100 amperes for 30 min. The gel bands were visualized under Ultra Violet (UV) illumination. A photomicrograph was taken using the gene-snap programme.
PCR product purification: PCR products were purified using QIAquick PCR purification Kit Protocol using a vacuum manifold (Qiagen). Briefly, 250 μL of Buffer PB was added to 50 μL of PCR product. To bind the DNA, the samples were loaded into the prepared QIAquick columns by pipetting and vacuum applied. After the sample had passed through the column, the vacuum source was switched off. To wash the DNA sample 750 mL of buffer PE was added to each QIAquick column and vacuum applied. Each QIAquick column was transferred to a 2 mL collection tube provided and centrifuged for 1 min at 10.000 x g. To remove residual ethanol present on the column, a second centrifugation step was required at 10.000 x g for 1 min. To elute the DNA, the QIAquick column was then transferred to a sterile 1.5 mL eppendorf and 40 μL of Buffer EB or Milli-q water added to the column and allowed to stand for 1 min, followed by centrifugation at 10.000 x g for 1 min.
DNA extraction from agarose gel: Following gel electrophoresis, the band of interest was excised using a scalpel, under a UV light. The DNA was extracted from the gel using the QIAquick Gel Extraction Kit, using a vacuum manifold. Buffer QG and PE were supplied by Qiagen. The DNA gel slice was weighed in a colourless tube and 3 volumes of Buffer QG was added to 1 volume of agarose gel (100 mg -100 μL) and incubated for a period of 10 min at 50°C. To help dissolve the gel, mixing by vortexing of the tube was done every 2-3 min during the incubation. After the gel slice had completely dissolved, 1 volume of isopranol was added. Following mixing, by gentle inversion, the mixture was applied to a MiniElute column and centrifuge at (10.000 x g) for 1 min. The flow-through was discarded and 500 μL of Buffer QG was added to the QIAquick column and a vacuum applied to remove residual agarose. Following centrifugation at (10.000 x g) for 1 min, the column was washed in 750 μL of buffer PE. A second spin followed to remove residual buffer. The QIAquick column was transferred to a clean 1.5 mL micro centrifuge tube, centrifuge for 1 min at 10.000 x g to remove residual ethanol (Buffer PE). The QIAquick column was placed in a clean 1.5 mL micro centrifuge tube and the DNA was eluted by adding 30 μL of Milli-Q water to the centre of QIAquick membrane and centrifuge the column for 1 min at 10.000 x g.
Insertion of foreign DNA into vector: The band intensities of the inserts and vector on gel were compared to determine the insert: vector molar ratio during ligation. DNA was diluted to the appropriate concentration with sterile Milli-Q water. The amount of insert needed to ligate 50 ng of vector DNA was calculated using the formula:
The ligation reaction was composed of 1 unit of T4 DNA ligase (Promega), 1 μL l of 10 x T4 DNA ligase buffer (Promega), 1 μL of pPIC9 vector and x μL of insert, where x μL gives an insert: vector molar ratio. Sterile Milli-Q water was added to give a final volume of 10 μL. The reaction was incubated for a period of 16 h at 4°C. 0.01 unit of T4 DNA Ligase is defined as the amount of enzyme required to catalyse the ligation of greater than 95% of the Hind III fragments of 1 μg of Lambda DNA at 16°C in 20 min.
Ligation with pETBlue-1 blunt vector: Ligation with pETBlue-1 blunt vector was done using the Novagen perfectly blunt cloning kits. This kit is designed by Novagen for simplified cloning of any DNA fragment regardless of whether the termini are blunt ends 5'-overhangs or 3'-overhangs. The PCR restricted digests was used as input DNA into this system. With the Perfectly Blunt Cloning Kits the target DNA is first converted to a blunt, phosphorylated form in a single brief reaction. Following a brief heat inactivation step, the blunt phosphorylated insert is combined with blunt dephosphorylated vector and then ligated. Subsequent transformation into the highly efficient NavaBlue competent cells generates recombinant colonies that are visualized easily by white/blue screening.
End conversion reaction: A reaction mixture was made with 1 μL PCR product, 4 μL nuclease free water and 5 μL of end conversion mix making a total volume of 10 μL. This was mixed gently by stirring with a pipette tip followed by incubation of end conversion reaction at 22°C for 15 min in the PCR machine. The reaction was inactivated by heating at 75°C for 5 min, then cooled briefly on ice for 2 min. The cooled reaction was centrifuged at 10.000 x g for 1 min before proceeding to the ligation reaction. The Ligation with perfectly Blunt pETBlue-1 vector required 1 μL (50 ng) Blunt vector and 1 μL (4U) T4 DNA ligase added to the End conversion reaction to make up a total reaction volume of 12 μL. This was incubated on the PCR machine at 22°C for 15 min.
Ligation of insert with pPIC 9 vector: The insert generated by cloning into pETBlue1 vector were ligated into digested pPIC 9 in the ration of insert/vector ratio of 1:2; 1:4 and 1:6 as shown on the Table 2.
Transformation into NovaBlue competent cells: The Novagen competent cells transformation protocol kits was used. The number of tubes of cells required were thawed on ice and gently mixed to ensure that the cells are evenly suspended. One microliter of the DNA solution was added directly to the cells and stirred gently to mix. The tubes were placed on ice for 5 min, followed by heating in a water bath at 42°C for exactly 30 sec. With no shaking the tubes were placed on ice for 2 min. Fifty microliter of room temperature SOC medium was added to each tube.
Plating of competent cells for growth: The Lauria-Berthani (LB) agar plates containing 50 μg mL-1 carbenicillin was poured on petri dishes under aseptic condition and allowed to set for about 30 min. Hundred microliter of transformation solution was transferred onto the centre of plate using sterile pipette tips. A bent glass rod plating spreader was completely immersed into ethanol and flamed to sterilized. After the flame is extinguished and allowed to cool for 10 sec prior to placing the spreader on the plate. The spreader was placed on the LB agar at the outside of the plate not touching the pool of cells, before spreading the cells. Slowly the plate was turned while supporting the weight of the spreader and cells spread until sample was evenly distributed on plate. The plates were allowed to sit upright at room temperature for 15 min prior to incubation. This allowed excess moisture to absorb into the plates before they were inverted and placed in the incubator at 37°C for 16 h, for cells to grow.
Selection of transformants: Five mL of LB solution were poured in 12 (50 mL) tubes, followed by the addition of 5 μL carbenicillin antibiotic. Each selected colonies (insert: vector ratio) contained 4 tubes. A forceps was flamed-sterilized and used to pick a sterile tooth pick that was used to pick a selected single bacterial colony from the LB agar plates. The colony was transferred to each LB solution and each tube labelled and placed into a shaker incubator at 37°C for 16 h, shaking at 250 rpm.
Purification of plasmid DNA: Plasmid DNA was extracted from cells using the QIAprep Spin Miniprep Kit. 1.5 mL of cells were pelleted by centrifugation at 10, 000 x g for 10 min. The supernatant was discarded and the pellet re-suspended in 250 μL of buffer P1. Cells were lysed by the addition of 250 μL of buffer P2. The resulting solution was inverted 4-6 times before an incubation period of 1 min. Cell lysis was stopped by the addition of 350 μL Buffer N3 and the tubes were gently inverted 4-6 times. Cell debris were removed by centrifugation at 14,000 x g for 10 min. The supernatant was transferred to a miniprep Spin Column and spun for 1 min at 10,000 x g. The column was washed by centrifugation (10, 000 x g) with 500 μL buffer PB, followed by a second wash with 750 μL buffer PE. A final spin was required to remove trace amount of ethanol. The Miniprep Spin Column was transferred to a sterile 1.5 mL eppendorf and an appropriate amount of Buffer EB or milli-q water (30-50 μL) was added. The DNA was eluted by centrifugation at 10,000 x g for 1 min.
Analysis of transformants: Transformants were screened by restriction endonuclease digestion. High copy plasmid DNA from 5 mL overnight culture of E. coli grown in LB medium was purified using the QIAprep spin columns on QIAvac 24.
Restriction endonuclease digestion of plasmid DNA: A double digestion of construct with SnaBI and XbaI restriction enzyme was used as shown in Table 3.
The reaction mix was incubated at 37°C for 2 h followed by PCR extraction described earlier, to remove SnaB1 and buffer before the second digest. After the second digest and further PCR purification agarose gel electrophoresis of 40 μL of purified plasmid construct with 10 μL loading dye was run on a gel loaded with 20 μL of 1.5 kb ladder and was ran at 200 v for 30 min.
Orientation of insert: Double restriction digest of insert with restriction enzymes BstE11 and Xba1, was done on purified plasmid DNA as shown on Table 4.
| Table 1: | Composition of standard PCR reaction product (50 μL reaction mix, assembled in a 0.5 mL PCR tube on ice) |
 | |
| Table 2: | Ligation of insert with pPIC9 at different concentration ratio |
 | |
| Table 3: | Double restriction digest of plasmid DNA insert with SnaB1 and Xba1 restriction enzyme |
 | |
| Table 4: | Double restriction digest for insert orientation using BstE11 and Xba1 enzyme |
 | |
After the restriction digest PCR extraction using QIAquick PCR purification kit protocol as described earlier was used to purified the plasmid DNA and a gel electrophoresis was used to obtain band for checking the orientation. Twenty microliter of insert was added 5 μL of loading dye and loaded along with a 10 μL (1.5 kb) ladder, then run at 200 v for 30 min.
Long term storage of transformed bacterial cells: Transformed E. coli cells were kept viable by preparing sterile 20% glycerol stocks. A single colony of transformed E. coli was inoculated into 5 mL of LB, containing the 5 μL carbenicillin antibiotic and incubated overnight at 37°C with vigorous shaking at 250 rpm. From this culture, 1 mL of cells were transferred to a sterile 1.5 mL eppendorf tube and spinned for 5 min. The supernatant was decanted and pellet resuspended in 1 mL 20% sterile glycerol and stored at -70°C.
RESULTS
Primer design: Three primers were successfully designed for the amplification of the Nucleotide Binding Domain (NBD1) of Sulfonylurea Receptor (SUR1) of hamster, with the nucleotide base pair within the 2003-2814 region. The NBD1 PCR fragments is summarized in Fig. 1. The forward primer of 24 –mer, with melting temperature Tm 69.6°C. The first reverse primer with his tag and polylinker of 72-mer and Tm (75°C). The second reverse primer with His tag had 51-mer with Tm of 75°C.
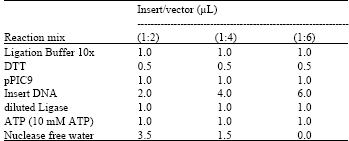
PCR amplification of NBD1: The KOD HiFi, Hot Start DNA Polymerase was used to successfully amplify the NBD1 of SUR1 from Hamster. The reaction conditions of denaturation for 20 sec at 98°C, annealing for 30 sec at 65°C, with no extension, of 30 cycles produced amplifications for both NBD1 with his tag and polylinker reverse primers. The bands were observed on agarose gel electrophoresis, under UV light. The two fragments with his tag and poly linker were amplified in the regions with 886 base pair and 907 base pair using a 50 base pair molecular marker as ladder on the agarose gel electrophoresis (Fig. 2).
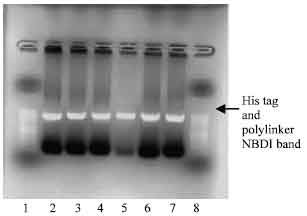
EcoRI restriction of NBD1 DNA and pPIC 9 cloning vectors: The NBD1 DNA gel extracted and purified and the pPIC 9 cloning vector were digested using the EcoRI restriction enzyme at 37°C incubation for 2 h. DNA of NBD1 with and without poly linker were digested by the EcoRI producing a fragment of 1307 base pair, but there was no restriction of the pPIC9 at first attempt, as shown on Fig. 3. Only the visible bands on agarose gel electrophoresis, for the digest DNA with his tag and poly linker were viewed under UV light.
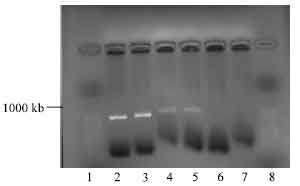
Optimization of pPIC9 vector restriction with EcoRI: Higher volumes of pPIC 9 vector (15, 20 and 25 μL) restriction condition were used to optimise the restriction reaction. Successful enzymatic restriction with EcoRI was observed for pPIC9 for 15 and 20 μL . The 20 μL produced stronger band intensities on agarose gel observed under UV light compared to the 15 μL . No band restriction was observed for 25 μL; therefore, the 20 μL volume was selected for the pPIC9 digest reaction as shown on Fig. 4.
Ligation of NBD1 digest with pPIC-9 vector: The first attempt to ligate the EcoRI digested insert directly into the EcoRI site of the digested pPIC-9 vector as described earlier, did not produce any colonies that would have indicated positive results.
 | |
| Fig. 1: | NBD1 DNA PCR fragments (includes restriction sites and 2 base additions for cutting) |
In the second ligation attempt very few colonies were produced on LB agar plate, but extraction and purification of the plasmid DNA from transformed colonies and analysis of the transformants produced no bands around the anticipated region on the agarose gel electrophoresis, using 50 base pair marker as observed under UV light. Due to the failure to ligate NBD1 insert into the pPIC-9 vector the pETBlue-1 blunt end vector which is smaller and has a higher ligation efficiency, was used for ligation with the NBD1 digest.
Ligation of NBD1 digest into pETBlue-1 blunt vector: To optimise ligation of NBD1 digest, the KOD HiFi DNA polymerase reaction was used to generate another amplified NBD1 DNA (Fig. 5). The quality of amplification product was necessary for direct cloning of PCR products without purification into pETBlue-1. The agarose gel electrophoresis reaction showed two clear distinct bands of the desired size for His tag and polylinker as observed under the UV light.
The end conversion reaction of the PCR mix produces blunt end amplified NBD1 inserts enhancing the possibility of successful ligation into the blunt end pPETBlue 1 vector. With the blunt end vector, the amplified insert did not require any EcoRI digestion before ligation and was transformed using Novagen competent cells. After transformation, the cells were cultured on LB agar plates in presence of 50 μg mL-1 carbenicillin antibiotics and incubated overnight. Five colonies of each plate of His tag insert and polylinker-his insert, were picked and grown in 5 mL LB overnight culture as described earlier. Plasmid DNA extraction followed by digestion with EcoRI was done to confirm success of ligation. As shown in Fig. 6, three of five colonies contained insert for plasmid DNA with his and only one colony out of five showed insert for the plasmid DNA with linker-His (Fig. 6)
There was only one colony of transformants with polylinker NBD1 plasmid DNA (Fig. 7).
 | |
| Fig. 2: | Photomicrograph of agarose gel electrophoresis of NBD1 DNA PCR amplification product. Lanes: 1 and 8= 50 base pair ladder; 2-4= NDB1 with His tag reverse primer; 5-7 = NBD1 with polylinker primer |
 | |
| Fig. 3: | Photomicrograph of agarose gel electrophoresis of restriction digest of DNA for NBD1 and pPIC 9 vector viewed under UV light. Lanes : 1 and 8= 50 base pair ladder; lanes 2-3= Restricted digest of DNA of NDB1 with His tag reverse primer; 4-5= NBD1 digest with poly linker primer, 6-7, pPIC9 no restriction observed |
 | |
| Fig. 4: | Photomicrograph of agarose gel electrophoresis of restriction digest of pPIC 9 vector viewed under UV light. Lanes: 1 and 8= 50 base pair ladder; lanes 2-3= digest at 15 μL l pPIC; 4-5= digest at 20 μL pPIC 9; 6-7= no digest at 25 μL pPIC 9 |
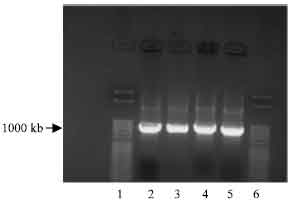 | |
| Fig. 5: | Photomicrograph of agarose gel electrophoresis of PCR amplification of NBD1 DNA used for perfectly blunt end cloning, viewed under UV light. Lanes: 1 and 6 = 50 base pair ladder; 2-3= NBD1 with His tag primer; 4-5= NBD1 DNA with polylinker |
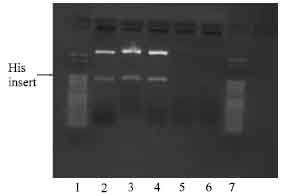 | |
| Fig. 6: | Photomicrograph of agarose gel electrophoresis of NBD 1 his tag, viewed under UV light. Lanes: 1 and 7 = 50 base pair ladder; 2-4 = restricted plasmid DNA of NBD 1 colonies with his tag primer; 5-6 = non restriction plasmid DNA colonies |
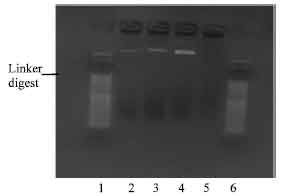 | |
| Fig. 7: | Photomicrograph of agarose gel electrophoresis polylinker NBD1 insert, viewed under UV light. Lanes: 1 and 6 = 50 base pair ladder; 2= plasmid DNA of NBD1 colonies; 3-5= non plasmid DNA colonies |
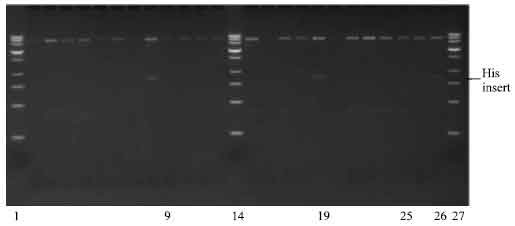 | |
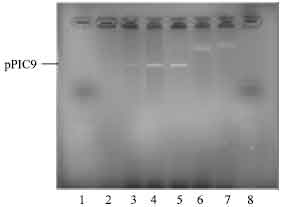
| Fig. 8: | Photomicrograph of agarose gel electrophoresis of transformed DNA construct after double restriction digest with SnaBI and XbaI restriction enzyme, viewed under UV light. Lanes: 1, 14 and 27 = 1.2 kilo base pair ladder; 9 and 19= insert from transformed colonies with vector/insert ratio of (1:4); 25-26= insert from transformed colonies with vector/insert ratio of (1:6) |
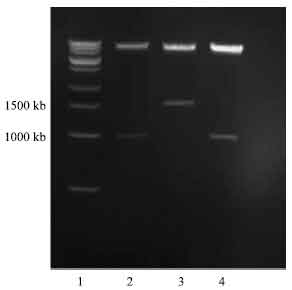 | |
| Fig. 9: | Photomicrograph of agarose gel electrophoresis of NBD1 DNA construct showing orientation of insert after double restriction digest with BstEII and XbaI enzyme, viewed under UV light. Lanes: 1 = 1.2 kilo base pair ladder; 2= correct orientation of insert with vector/insert ratio of (1:4); 4= correct orientation of insert with vector/insert ratio of (1:6) |
The band on lane 2 was very faint indicating low concentration of plasmid DNA insert.
Cloning of insert into pPIC 9 vector: The pETBlue-1 construct with transformed NBD1 His and poly-linker inserts were digested as well as pPIC9 with EcoRI, creating sticky ends to the multi-cloning sites, that will accept the EcoRI digest inserts. Plasmid DNA purification of insert with His tag was done as described earlier. Ligation of pPIC 9/insert ratio was (1:2; 1:4 and 1:6). The ligation was carried out as described earlier and followed by transformation into Competent NovaBlue cells. The ligation was carried out on PCR machine incubated at 4°C for 3 days, instead of the standard 24 h at 16°C incubation. The transformation yielded many colonies. A total of 12 colonies were picked for the NBD1 insert (4 colonies per insert/vector ligation ratio). The plasmid DNA purified were digested with SnaBI and XbaI restriction enzyme, to determine whether the ligation was successful. Out of the 12 colonies selected, only 3 (one with the insert/vector ration of 1:4 and 3 with insert/vector of 1:6) contained the His/NBD1/pPIC-9 construct (Fig. 8). The rest of the pPIC9 vector re-annealed during ligation and was transformed into the competent cells. Agarose gel electrophoresis of purified plasmid construct showed the presence of insert for some transformed colonies with bands that lies around the 1300 base pair region.
Double digest of insert for checking the orientation: The correct orientation of insert was important in order for the expression of the NBD1 plasmid DNA into the yeast cell (Pichia pastoris). The double restriction digest was done to check the direction into which the insert was ligated into the vector, using BstEII and XbaI restriction enzymes at 60 and 37°C incubation for 2 h, respectively followed by successive PCR extraction. The correct orientation was observed for two of the three inserts (Fig. 9). The correct orientation was determined by observation of two bands. One band for digested insert with 1300 base pair and a larger band at 8000 bp. For the wrong orientation the smaller insert contains 1700 bp and the larger vector band would be around 7500 bp as shown in Fig. 9.
DISCUSSION
The NBD1 nucleotide of SUR1 was successfully amplified by the KOD HiFi DNA polymerase reaction. The fidelity of the amplification reaction was positively assessed by cloning. The KOD HiFi DNA polymerase is a recombinant form of Thermococcus kodakaraensis KOD 1 DNA polymerase[23]. KOD HiFi is a very high fidelity thermostable polymerase, amplifying target DNA up to 6 kilo base pair (kbp), with superior accuracy and yield[4]. The enzyme’s 3'-5'exonuclease dependent proofreading activity results in a lower PCR mutation frequency than any other commercially available DNA polymerase. The elongation rate and progressivity are 5 times and 10-15 times higher, respectively, than Pfu DNA polymerase resulting in higher accurate and robust yield in a short time. KOD HiFi DNA polymerase produces blunt DNA products and that made it possible for the cloning of NBD1 amplified products in Novagen’s Perfectly Blunt pET Blue-1 vector.
There has been no early attempts made to amplify and clone the nucleotide binding domains of SUR1 from Hamster for the K-ATP insulin regulation studies. The successful amplification and cloning of the NBD1 into a perfectly blunt vector and subsequently into a larger pPIC-9 vector is a significant progress towards the expression, purification and subsequent structural studies of the nucleotide binding domains in SUR1.
The Perfectly Blunt Cloning strategy was adapted only after the first attempt of direct cloning of NBD1 insert into the pPIC-9 vector failed. The Blunt cloning therefore simplified cloning of the amplified DNA generated by PCR using any type of DNA polymerase[24]. With the Perfectly Blunt Cloning, it is also possible to go from PCR product to plating transformants in less than 1 h with minimal hands-on time[25].
The pETBlue-1 used for the cloning of NBD1 digest facilitated the expression of unfused, native protein from an insert that provided an ATG start codon. The blunt cloning site is located just downstream from the T7 gene 10 RBS (ribosome binding site). Amplification with sense primers beginning with ATG at their 5' end will ensure optimal spacing between the RBS and translation initiation sites for efficient protein synthesis in E. coli. However, the sense primer for NBD1 amplification and cloning was designed without the ATG start codon, to suit expression in yeast Pichia pastoris, therefore with the absence of start codon ATG expression into E. coli was not possible[4].
The pPIC-9 vector used for the cloning of the NBD1 insert is a fusion vector with multiple (XhoI, SnaBI, EcoRI, AvrII, NotI) unique restriction sites[26]. It is an ideal cloning tool when strong, highly inducible promoter for protein expression is utilized. It is also ideal for secreted expression protein using the α-factor secretion signal into Pichia pastoris[27,28].
The Novagen Competent Cells used for transformation of the NBD1 insert enabled a convenient, efficient construction of plasmid recombinants. The cells are grown and made chemically competent by an optimised procedure, followed by verification of cloning efficiency and strain identity[29,30]. Experimental DNA has been shown to produce no colonies or very low number of colonies but the test plasmid included in the kit (Novagen) yields expected efficiency. This happens when the experimental DNA contains an inhibitor of ligation (excess salts, EDTA, proteins, etc) that can inhibit ligation or contains inhibitor of transformation[31,32]. No colonies or low copy number of colonies may suggest that the vector and /or insert have been damaged or otherwise have incompatible ends. The cloning strategy was changed after the transformation of the NBD1 insert into pPIC-9 vector failed. The vector insert ratio was optimised and the use of fresh, reliable reagents for DNA preparation. The amplified NBD1 PCR products was faster to first clone them using a Perfectly Blunt pETBlue-1 vector, before ligating into the pPIC-9 vector using EcoRI restriction enzyme to excise the fragment.
Cloning and subsequent expression, purification and crystallization of the nucleotide binding domains of SUR1 would enhance the understanding of ß-cell K-ATP channel, which is a multimeric complex composed of two different types of subunits: a pore-forming subunit, Kir6.2 and a regulatory subunit, the sulfonylurea receptor SUR1[1,33]. Both subunits are required for functional K-ATP channels since expression of Kir6.2 alone does not result in measurable currents.
Studies, however, have shown that removal of the last 26 amino acids of Kir6.2 (Kir6.2CΔ26) enables functional expression of the protein in the absence of the SUR1[5]. This enables those K-ATP channel properties that are intrinsic to the Kir6.2 subunit and those that are conferred by association with sulfonylurea receptor to be determined. Such studies have shown that the site at which ATP and ADP mediate channel inhibition lies on Kir6.2, whereas SUR1 confers sensitivity to the sulfonylureas, diazoxide and the potentiatory effects of MgADP[8,34].
SUR1 is an ATP-binding cassette transporter protein[16,17,35] and has two nucleotide-binding domains (NBDs) that contain highly conserved Walker A (WA) and Walker B (WB) consensus sequences. In other ATP-binding cassette transportes and ATPases, these motifs are involved in the binding and hydrolysis of ATP[17,35]. Two residues are of particular importance: an aspartate residue within the Walker B motif coordinates the Mg2+ ion of MgATP, while lysine in Walker A motif is critical for ATP hydrolysis. In SUR1 these residues are involved in the ability of MgADP to potentiate K-ATP channel activity[36,37].
Cloning and expression of an inwardly rectifying ATP-regulated potassium channel from rat kidney has been reported[38]. A complementary DNA encoding an ATP-regulated potassium channel was isolated by expression cloning from rat kidney. The predicted 45K protein, which features two potential membrane-spanning helices and a proposed ATP-binding domain, represents a major departure from the basic structural design characteristic of voltage-gated and second messenger-gated ion channels. However, the presence of an H5 region, which is likely to form the ion conduction pathway, indicates that the protein may share a common origin with voltage-gated potassium channel proteins[16,22,38].
Several derivatives of 3-phenylpropionic acid (e.g. nateglinide) or benzoic acid (e.g. meglitinide and more lipophilic repaglinide) inhibit KATP-channels by binding to the same receptor site that mediates the responses to sulfonylureas[39-41]. Two of these sulfonylurea analogues (repaglinide and nateglinide) have recently been introduced into the therapy of two type 2 diabetes mellitus[5,15,42].
This project is of significance because of the importance of ATP-sensitive K (KATP) channels in transmembrane transport in muscle cells, neurons and most other excitable cells. These channels serve to adjust the resting membrane potential to the metabolic state of the cell[1,17] The K (KATP) channels are the targets for a number of drugs that can block them, like the sulfonylureas such as tolbutamide, or open them like cromakalim, pinacidil, or diazoxide (sulphonamide)[19,43]. These drugs are of great therapeutic interest because they provide a way to pharmacologically adjust the excitability of cells, raising it with blockers and lowering it with openers[44]. Widespread use of potassium channel openers (KCOs) has been shown to be impaired by their poor tissue specificity and low affinity[45-47]. In the search for better drugs species in terms of specificity, it is necessary to understand the mechanism of action and the 3-D structure of the regulatory signal proteins, most especially the nucleotide binding domain regulatory subunit SUR1 of the KATP channels.
The cloning of NBD1 into the pPIC-9 expression vector has been achieved with the use of KOD HiFi DNA Ploymerase which successfully amplified the gene encoding the NBD1 of sulfonylurea receptor (SUR1). SUR1 inwardly rectifying subunit of the KATP sensitive channel plays an important role in insulin regulation and diabetes therapy. The cloning of NBD1 is a significant achievement towards the SUR1 project. With the cloned NBD1 in the right orientation, progress can now be made towards the expression, purification, assay development and 3-D structure determination. The global success of this project could be a major breakthrough in the drug discovery projects related to insulin regulations and also in diabetic therapeutic innovations.
REFERENCES
- Proks, P., F. Reimann, N. Green, F. Gribble and F. Ashcroft, 2002. Sulfonylurea stimulation of insulin secretion. Diabetes, 51: S368-S376.
CrossRefPubMedDirect Link - Walker, J.E., M. Saraste, M.J. Runswick and N.J. Gay, 1982. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1: 945-951.
CrossRefDirect Link - Tsuchimoto, D., Y. Sakai, K Sakumi, K. Nashioka, M. Sasaki, T. Fujiwara and Y. Nakabeppu, 2001. Human APEZ protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APEZ2 is partly associated with proliferating cell nuclear antigen. Nucl. Acid Res., 29: 2349-2360.
- Studier, F.W. and B.A. Moffatt, 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol., 189: 113-130.
CrossRefPubMedDirect Link