
Abbas Bahador
Department of Microbiology, Faculty of Medical,
Tehran University of Medical Sciences, Tehran, Iran
Hormozdyar Etemadi
Department of Microbiology, Faculty of Medical,
Tehran University of Medical Sciences, Tehran, Iran
Bahram Kazemi
Department of Molecular Biology
Asian Network for Scientfic Information Shahid Beheshti University of Medical Sciences, Tehran, Iran
Roghayeh Ghorbanzadeh
Department of Microbiology, Faculty of Medical,
Tehran University of Medical Sciences, Tehran, Iran
Journal of Medical Sciences
Year: 2004 | Volume: 4 | Issue: 4 | Page No.: 252-256







ABSTRACT
Polymerase Chain Reaction (PCR) has been widely used due to its high specificity, sensitivity and rapid turn-around time. However, inhibitory factors may be co-extracted with the target nucleic acid that will hinder the performance of PCR. The major difficulty with mycobacteria is achieving optimal cell lysis. Due to a genuine need for an accurate test for the diagnosis of tuberculosis a comparison of DNA extraction methods was conducted. DNA extraction methods for Mycobacterium tuberculosis were evaluated including Triton, Chelex, Nonidet, SDS/Lysozyme and Silica-based methods on bacterial cells and spiked sputum. DNA extracted from the above procedures was diluted from 10-1 to 10-7 for use as templates in the PCR titration assay. The PCR end point was at a dilution of 10-2 when DNA was extracted using the Triton and Chelex extraction methods. It was 10-3 for Nonidet and 10-4 for SDS/Lysozyme extraction methods. DNA extractions by silica-based method had PCR end point titrations at the 10-5 dilution. The sensitivity of extraction by silica-based is 1-10 cells. In the present study silica-based method is effective extraction method. The end point titrations are identical for bacterial cells and spiked sputum. Inhibition was not observed in PCR with DNA isolated from spiked sputum. Silica-based method required the least labor and completion time
PDF Abstract XML References
How to cite this article
Abbas Bahador, Hormozdyar Etemadi, Bahram Kazemi and Roghayeh Ghorbanzadeh, 2004. Comparison of Five DNA Extraction Methods for Detection of Mycobacterium
tuberculosis by PCR. Journal of Medical Sciences, 4: 252-256.
DOI: 10.3923/jms.2004.252.256
URL: https://scialert.net/abstract/?doi=jms.2004.252.256
DOI: 10.3923/jms.2004.252.256
URL: https://scialert.net/abstract/?doi=jms.2004.252.256
INTRODUCTION
In microbiology, DNA amplification using PCR has allowed great progress to be made in the rapid and accurate diagnosis of infections due to organisms that are not cultivable by in vitro means, that require complex media or cell cultures and prolonged incubation times, or for which culture is too insensitive. Amplification techniques for the diagnosis of tuberculosis have attracted considerable interest, particularly with the hope of shortening the time required to detect and identify Mycobacterium tuberculosis in respiratory and nonrespiratory specimens[1,2]. The major inconvenience of PCR is the presence of inhibitors, which interfere with amplification-based techniques. The major difficulty with mycobacteria is achieving optimal cell lysis. Commercial kits have been developed to allow the majority of clinical laboratories access to amplification-based techniques[3-6], but they have perhaps oversimplified such techniques and more precisely, the sample preparation and DNA extraction steps. The buffers used in commercial kits do not allow complete mycobacterial cell lysis[7]. Due to a genuine need for an accurate test for the diagnosis of tuberculosis in patients, a comparison of DNA extraction methods was conducted.
MATERIALS AND METHODS
Cell stock suspension: Mycobacterium tuberculosis (Mtb) isolated from 5 patiants were grown in Middlebrook 7H10 broth at pH 7.0 supplemented with 2% Middlebrook ADC, 0.5% Tween 80, 0.04% Mycobactin J and incubated at 35°C for 2-3 weeks. Cells were harvested by centrifugation at 1500 g for 15 min. The pellet was washed twice with 5 mL of Phosphate-buffered Saline (PBS), pH 7.2 (PBS) and adjusted to a concentration of 2.0x104-105 cells mL-1 based on the value of 1 OD550 nm = 2.8x106-107 cells mL-1.
Spiked sputum: Sputum samples were screened for Mtb by direct smear and culture. Remainders of the sputum samples were stored at -70°C until the culture results were determined. Five of these samples that tested negative for Mtb were thawed and thoroughly mixed to provide a homogeneous sample matrix for any extraction method. Five milliliter of the NALC(N-Acetyl L-Cystein)-NaOH solution was added to 5 mL of sputum samples in 50 mL conical tubes (final NaOH concentrations in the samples is 3.0%) vortexed and incubated at room temperature for 15 min. The samples were then diluted to the 50 mL mark with 0.067 M phosphate buffer pH 6.8, mixed by inversion and centrifuged for 15 min at 3000 g. After centrifugation, the supernatant is decanted and the pellet is resuspended in 1 mL 0.2% normal saline[8]. The sputum was mixed with 1 mL of cell stock suspension (2.0x104-105 cells mL-1) and diluted to a final volume of 5 mL with PBS, pH 7.2 (spiked sputum). This mixture was extracted by the different methods described below:
DNA extraction methods: Fifty microliter of the cell stock suspension containing 2.0x104-105 cells mL-1 were added to 200 μL of PBS, pH 7.2 (working cell suspension) and was used for all experiments in the study. A 250 μL aliquot of the spiked sputum was used. mycobacteria was lysed in the working cell suspension and spiked sputum using the following methods.
Triton method: Two hundred and fifty microlitre of lysis buffer (100 mM NaCl, 10 mM Tris-HCl, pH 8.3; 1 mM EDTA, pH 9.0; 1% Triton X-100) was added to an equal volume of working cell suspension or spiked sputum. Both suspensions were boiled for 15 min and allowed to cool to room temperature[9].
Chelex method: Two hundred and fifty microlitre of working cell suspension or spiked sputum were extracted by adding 200 μL 5% Chelex-100 resin (Bio-Rad) and 4 μL proteinase K (10 mg mL-1). The mixture was incubated at 56°C for 45 min and for 8 min at 100°C[10].
Nonidet method: Two hundred and fifty microlitre of lysis buffer (50 mM KCl, 50 mM Tris-HCl, pH 8; 2.5 mM MgCl2, 0.45% Tween20, 0.45% Nonidet P-40 and 100 μg of Proteinase K mL-1) was added to an equal volume of working cell suspension or spiked sputum. Both suspensions were incubated at 56°C for 3 h. Proteinase K was inactivated by 15 min of incubation at 95°C[11].
SDS/Lysozyme method: Two hundred and fifty microlitre of specimens were resuspended in a final volume of 1 mL of Tris-EDTA, pH 7.6 containing 10 mg of lysozyme mL-1. Samples were then incubated at 37°C for 1 h; 30 μL of proteinase K (14 mg mL-1) and 3% SDS were added, followed by incubation for 2 to 3 h at 56°C or overnight at 37°C. Proteinase K was inactivated by 15 min of incubation at 95°C[7].
Silica-based method: Lysis buffer was prepared by dissolving 120 g of guanidine thiocyanate (Fluka Chemie AAG, Switzerland) in 100 mL of 0.1 M Tris-HCl, pH 6.4, followed by addition of 22.0 mL of 0.2 M EDTA, pH 8.0 and 2.6 g of Triton X-100. One milliliter of the lysis buffer was added to 250 μL of working cell suspension or 250 μL of spiked sputum, followed by 40 μL of acid-washed silica. The suspension was vortexed for 10 s and incubated for 15 min at room temperature with constant mixing. The silica was pelleted by centrifugation at 13,000 g for 30 s. The supernatant was discarded and the pellet was washed twice with buffer containing 12% guanidine thiocyanate in 0.1 M Tris-HCl, pH 6.4, followed by two washes of 70% ethanol and one of acetone. The pellet was dried at 56°C for 5 min and then reconstituted in 50 μL of 12 mM Tris-HCl, pH 7.4[12].
Following Triton, Chelex, Nonidet and SDS/Lysozyme cell lysis, complete DNA purification (cetyltrimethylammonium bromide [CTAB]-Roche) was performed in an identical manner for all samples. The 500 μL aliquot was treated as previously described[13]. Briefly, the samples were incubated in a solution of CTAB-NaCl (50 μL of 5 M NaCl and 40 μL of 10% CTAB) for 10 min at 65°C and then mixed with an equal volume of chloroform-isoamyl alcohol (24:1 [v/v]; 700 μL) and centrifuged for 15 min at 13,000 g in an Eppendorf centrifuge. The aqueous phase (650 μL) was then separated and mixed with an equal volume of isopropanol. The samples were left at -20°C for 30 min and then centrifuged for 15 min at 13,000 g. The DNA pellet was washed once with 70% ethanol, the pellet was dried at 56°C for 5 min and solublize pellet in 50 μL TE buffer[7]. DNA extracted from the above procedures was diluted from 10-1 to 10-7 for use as templates in the PCR titration assays.
Determination of sensitivity: Cell stock suspension containing 2x104-105 cells mL-1 was diluted from 10-1 to 10-4. One milliliter of each dilution was added to 1 mL of sputum followed by thorough mixing. PBS, pH 7.2 was added to a final volume of 5 mL and the suspensions were mixed again. DNA from a 250 μL aliquot of each of the spiked sputum was extracted for any extraction methods experiments and the DNA was eluted in a final volume of 50 μL. Likewise, 50 μL aliquots of the 10-1 to 10-4 dilutions of the cell stock suspension were extracted as above to compare the efficiency of the extraction procedure in cell versus spiked sputum .
Construction of an Internal Control (IC): As an internal control, an 81-bp fragment of the Ki-ras gene was amplified by PCR using primers KR-1 and KR-17 For Ki-ras gene:
KR-1: 5'-GGCCTGCTGAAAATGACTGA-3'
KR-17: 5'-TAGCTGTATCGTCAAGGCAC-3'[14].
The PCR product was analyzed on a 1% agarose gel and the IC band was cut out and eluted from the agarose using the gel extraction. The IC product was used as a template with Mtb PCR primers to ensure that no other bands were formed. The amount of IC to be added to each PCR reaction was optimized by titration with Mtb DNA to ensure it did not interfere with target Mtb DNA amplification.
PCR: A set of mycobacterium-specific primers (MF, 5'CGACCACTTCGGCAACCG3'; MR, 5'TCGATCGGGCACATCCGG3') was used to amplify rpoB DNA (342 bp) encompassing the Rifr region, which is associated with rifampin resistance in M. tuberculosis. The primers were selected from the highly conserved regions on the basis of known rpoB sequences. Template DNA and 20 pmol of each primer were added to a PCR mixture tube which contained 1U of Taq DNA polymerase, 250 μM each deoxynucleoside triphosphate, 50 mM Tris-HCl (pH 8.3), 40 mM KCl, 1.5 mM MgCl2 and gel loading dye and the volume was adjusted to 20 μL with distilled water. The reaction mixture was subjected to 30 cycles of amplification (30s at 95°C, 30s at 60°C and 45s at 72°C) followed by a 5 min extension at 72°C[15].
RESULTS AND DISCUSSION
Comparison of extraction methods for working cell suspension and spiked sputum : The PCR end point was at a dilution of 10-2 when DNA was extracted using the Triton and Chelex extraction methods. It was 10-3 for Nonidet and 10-4 for SDS/Lysozyme extraction methods. DNA extractions by Silica-based had PCR end point titrations at the 10-5 dilution. The efficiency of DNA extraction by silica-based was higher than the other methods (Table 1). Extractions using the spiked sputum exhibited no inhibition with all extraction methods. The internal control enabled the detection of the inhibition. There was no significant difference in the results of the PCR end point titrations between cell stock suspension and spiked sputum by each of the different extraction methods. An 81-bp fragment of the Ki-ras gene was uniformly amplified from all of the samples.
Comparison of sensitivity: The sensitivities of PCR using DNA extracted by the Silica-based method from the 10-1 to 10-4 cell stock dilutions and the spiked sputum prepared were compared. Based on the starting concentration, the number of Mtb cells extracted were 100-1000, 10-100, 1.0-10 and 0.1-1.0 and the sensitivity of cell stock dilutions and the spiked sputum were 1-10 cells.
| Table 1: | Summary of PCR end point titration with working cell and spiked sputum samples. Average result of replicates using same pooled sputum |
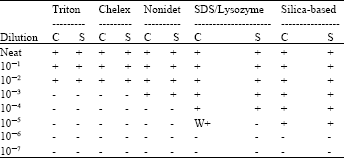 | |
| C = cells, S = spiked sputum, + = PCR positive. W+ = weak positive, - = PCR negative | |
| Table 2: | Comparison of different methods for DNA extraction based on an average of five samples |
 | |
The extraction methods were also compared with the ease of extraction, labor and completion time (Table 2). All extraction methods are fairly easy to perform, but the hands-on time is slightly different from one to another. Silica-based has the shortest time of completion. Since the cost of labor is the most expensive component, consideration has to be given in choosing the simplest protocol with the least labor and completion time.
A number of unanswered questions regarding the development of molecular techniques in the clinical laboratory for the diagnosis of tuberculosis have yet to be addressed: (I) which method should be used for extraction of mycobacterial DNA, (ii) should a positive internal control always be used with molecular methods of this sort and specifically, should this control be added to the sample preparation at the very start of the process to determine the efficacy of sample preparation and amplification procedure[1,16]. The aim of present study was to try to answer of these questions. We specifically chose the extraction methods to examine spiked sputum, for which amplification techniques are of considerable interest. The PCR assay used in this study targets rpoB sequences. In the study, DNA was extracted from approximately 1000-10,000 cells by using Triton, Chelex, Nonidet, SDS/Lysozyme and Silica-based cell lysis. By making 10-fold serial dilutions of the DNA before amplification, the end points of detection was 10-5 dilution for silica-based, indicating the efficiency of extraction in this method. The end point PCR for triton and chelex extraction was two log and nonidet one log lower than the silica-based methods. In the presence of sputum material, the PCR end point titration of each of the extraction methods was the same as in the absence of sputum material (Table 1), however, further testing using different sputum samples is necessary to confirm whether this observation is consistent regardless of variations in sputum matrices. The need for dilution is detrimental, especially when the bacterial load is low in clinical samples and in the presence of inhibitors. With working cell suspension and sputum material, the sensitivity of extraction by silica-based using end point PCR titration is at the level of 1-10 cells. Tell et al.[17] compared four rapid DNA extraction techniques for conventional PCR testing of three Mycobacterium spp. (M. avium, M. genavense and M. fortuitum) that affect birds. DNA extraction methods included enzymatic lysis, boiling followed by enzymatic lysis, freezing and thawing followed by enzymatic lysis and bead beating followed by enzymatic lysis. The bead beating with enzymatic lysis technique yielded significantly purer and higher concentrations of extracted DNA compared with other DNA extraction methods. Chui et al.[18] evaluated DNA extraction methods (rapid lysis, organic extraction, silica-based and magnetic particle-based [MagaZorb] technologies) for Mycobacterium avium subsp. Paratuberculosis. Efficiency of the extraction was determined by PCR. MagaZorb proved to be more efficient. Caldarelli-Stefano et al.[19] demonstrated that Magnetic bead DNA extraction can be used on both frozen and archival tissues, the method was reliable, simple, sensitive and rapid, in addition, it did not use hazardous procedures or specialised laboratory equipment and could be used for routine DNA isolation from various human tissues. The labor required to extract DNA differs slightly from one method to the other. The completion time for extraction using Nonidet and SDS/Lysozyme extraction was similar, but these two methods took longer than the Triton, Chelex and Silica-based methods. Generally, labor is the most expensive component of extraction procedures; therefore, great consideration has to be given to the protocol that requires the least "hands-on" time. The silica-based is as expensive as the other assays, however, with the shortest "hands-on" time, extraction with the silica-based is the most efficient and cost-effective way of performing DNA extraction for Mtb PCR.
REFERENCES
- Ieven, M. and H. Goossens, 1997. Relevance of nucleic acid amplification techniques for diagnosis of respiratory tract infections in the clinical laboratory. Clin. Microbiol. Rev., 10: 242-256.
Direct Link - Soini, H. and J.M. Musser, 2001. Molecular diagnosis of mycobacteria. Clin. Chem., 47: 809-814.
Direct Link - Boom, R., C.J. Sol, M.M. Salimans, C.L. Jansen, P.M. Wertheim-van Dillen and J. van der Noordaa, 1990. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol., 28: 495-503.
PubMedDirect Link - Honore, S., J.P. Vincensini, L. Hocqueloux, M.E. Noguera, D. Farge, P.H. Lagrange and J.L. Herrmann, 2001. Diagnostic value of a nested polymerase chain reaction assay on peripheral blood mononuclear cells from patients with pulmonary and extra-pulmonary tuberculosis. Int. J. Tuberc. Lung Dis., 5: 754-762.
- Kim, B.J., S.H. Lee, M.A. Lyu, S.J. Kim and G.H. Bai, 1999. Identification of Mycobacterial Species by Comparative sequence analysis of the RNA polymerase gene (rpoB). J. Clin. Microbiol., 37: 1714-1720.
PubMedDirect Link