
Al-Said Haffor
Associate Professor of Physiology
Depaltment of Zoology
College of Science
P.O. Box 2455
King Saud University
Riyadh 11541, Saudi Arabia
Journal of Medical Sciences
Year: 2004 | Volume: 4 | Issue: 2 | Page No.: 170-175







ABSTRACT
Obstructive sleep apnea (OSA) relates to cardiovascular diseases. This study evaluated oxygenation and the ventilatory responses to hypercapnea in OSA in whom cardiac output is elevated. Sleep studies were conducted using CASE system. From an arterial line, blood pressure was monitored directly by blood pressure transducer and blood samples were drawn from the same arterial line for arterial blood gases (ABG) analysis. Cardiac output was calculated by the indirect Ficks` method. Gas exchange measures were determined using automatic Douglas Bag System in which expired gases were analyzed online using mass spectrometer (Perkin Elmer-1100). Ventilation was recorded continuously by mass flow meter (TSI). Results of sleep studies indicated that the average obstructive apnea index was 74%. During wake state, the average cardiac output was 9.86 LPM, alveolo-arterial PO2 differences (A-a PO2) was 18.82 Torr and normalized VE was 3X for each level of CO2. At maximum respiratory response PaCO2 was elevated reflecting increased capacity for CO2 stores leading to increased CO2 load on the lungs. Present data indicated that significant obstructive sleep apnea occurs in adult in whom cardiac output, heart rate and blood pressure were elevated during day time. Based on the results of the present study it can be concluded that OSA is considered a risk factor for heart diseases.
PDF Abstract XML References Citation
How to cite this article
Al-Said Haffor, 2004. Obstructive Sleep Apnea in Elevated Cardiac Output Patients: Hypercapnea-induced Hypoapnea. Journal of Medical Sciences, 4: 170-175.
DOI: 10.3923/jms.2004.170.175
URL: https://scialert.net/abstract/?doi=jms.2004.170.175
DOI: 10.3923/jms.2004.170.175
URL: https://scialert.net/abstract/?doi=jms.2004.170.175
INTRODUCTION
Sleep apnea is classified into obstructive, central and mixed types. Obstructive sleep apnea (OSA) is characterized by repetitive intermittent obstruction of the upper airway during sleep, whereas central sleep apnea is characterized by recurrent apnea episodes in the absence of upper airway obstruction[1]. In OSA, the temporal failure of the pharynx to dilate on inspiration during sleep leads to an instantaneous cessation of breathing and can be a major cause of transient hypercapnea. These episodic ventilatory insufficiencies cause intermittent asphyxia, chemoreceptor stimulation, increased respiratory efforts with less effective ventilation and transient arousals[2].
Ventilation and circulation are vital and essentially autonomous biological functions that change in a direction-coupled manner to meet metabolic demands. This direction-coupled could be reversed in OSA[3-5]. Although results of many studies indicated that OSA is an important risk factor for systemic hypertension, myocardial infarction, stroke and sudden death[6-11] however, the mechanisms that tie this relationship have not been fully understood. Information regarding OSA responsiveness to hypercapnea has not been reported. Failure of the ventilatory system to match circulatory compensation result in failure in adjusting CO2 clearance by the lungs to metabolic production leading to increased in CO2 stores[12,13] which in turn increase the CO2 load on the lungs leading to increased pulmonary blood volume, via loco vasodilatation mechanism. It has been shown that, weeks of episodic hypoxia in rats led to chronic systemic and pulmonary hypertension after the hypoxemia was removed[14]. Moreover even in the healthy human, systemic blood pressure rises during stay in the hypoxia of high altitude and remains elevated for several days upon return to sea level[15-17]. It was reported that, increased sympathetic nerve activity is obligatory to the acute pressor responses to apnea[18], which elevates systemic resistance and blood pressure. On the other hand, transient elevations in systemic blood pressure have been shown to depress ventilation and to increase pharyngeal collapsibility[19]. Thus, the possibility that the acute and chronic hypertension caused by OSA can in turn exacerbate OSA in positive feed forward mechanism, in attempt to compensate. Because of the neural input from lung and chest wall receptors is different during periods of transient hypoxia as compared with periods of hypercapnea during which the upper airway is obstructed rather than opened[9]. In this regard hypercapnea stimulus elicits arousal response and termination of apnea. Information regarding the ventilatory response to hypercapnea in OSA patients has not been reported. The purpose of this study was to evaluate the ventilatory responses induced by hypercapnea in OSA in whom cardiac output is elevated. The second purpose was to demonstrate and introduce a steady state hypercapnea-induced hyperapnea test as a simple noninvasive and cost effective test to rule out risk for heart disease in OSA patients.
MATERIALS AND METHODS
General method: Six male patients diagnosed as OSA participated in the study. All signed approved consent form that was approved from the Institutional Review Board of LAC+USC Medical Center, Los Angeles, CA, USA. Metabolic measurements were obtained using Automatic Douglas Bag System[3] in which gas analysis was analyzed with an automated on-line mass-spectrometer (Perkin Elmer- 1100). From a brachial arterial line blood pressure was monitored directly by BP transducer and blood samples were drawn from the same line for arterial blood gas (ABG) analysis.
Sleep apnea procedure: Sleep studies were conducted using computer aided sleep system (CASS)-CNS POLYGRAPH. The system used MONTAGE file management to collect, store and analyze analog data acquisition. The software records and compare respiratory events such as air flow, chest and abdominal movements, eye movement, EEG, O2 saturation, PETCO2, FECO2. All measures were compared in accordance with the criteria of the national commission on sleep disorders research (NCSDR) to determine apnea index.
Cardiovascular measurement: Cardiac output was measured by rebreathing equilibration technique as described by Haffor and Nail[3]. Three trials were conducted with about 10 -15 min elapsed between trials to eliminate carry over effects of CO2. The average value was calculated for the three trials to give indication of day time cardiac output over a period of 1-2 h.
Arterial blood gas sampling: Following local anesthesia, a catheter was placed in the radial artery that was interfaced with a transiducer for measuring real time tension time index, blood pressure and heart rate. The catheter was inserted using Seldinger technique in which the artery is first located by palpation, anesthesia of the site was obtained by infiltrating 1 ml of lodicane. The needle with the obdurate half-inserted was the passed through the skin and advanced toward the artery at an angle about 60°. Once the wire guide is up to 10 cm within the artery, the needle was slowly withdrawn over it and was replaced by a shaped polyethylene. The catheter was filled with heparin-saline solution and taped to the skin and the puncture site is covered with gauze and a bandage. A 100 μl glass syringe was used to withdraw arterial blood for analysis of blood-gas status. A 80-100 μl sample of arterial blood was drawn into the syringe and immediately analyzed on a blood-gas analyzer (ABL Instrumentation Laboratory).
Hypercapnea-induced hyperapnea procedure: A modified steady state CO2 response for all patients were measured via open circuit method in which various concentrations, 0-8% CO2 in inspired air, was breathed until a steady state in PETCO2 was reached. Measurements of PET CO2, PaCO2, P100, HR, BP and VE were made once steady state was established for each concentration.
RESULTS
Results revealed that, the maximum breathing capacity (MBC) was severely impaired which was about 50% of normal values. The group also showed tendency for obesity of 35% fat as compared with 25% for average population values for this age group (Table 1).
Evaluation of oxygenation during sleep: Table 2 showed that the obstructive apnea index was 76%, 10% mixed index and 11% central index. Based on NCSDR criteria for OSA are met in this group[1]. Therefore, patients engaged in the study are diagnosed with OSA with an average SaO2 of 85% during sleep as compared with 91% during wake state (Table 3).
Evaluation of oxygenation during awake: As shown in Table 3, the partial pressure of oxygen in arterial blood (PaO2) , the alveolar to arterial oxygen difference (A-a PO2) and dead space to tidal volume ratio. During awake, oxygenation of this group is somewhat adequate as evident by 80% of PaO2 to PAO2 ratio at room air. However, the average A-a PO2 is 19 Torr, which reflect about 20% venous add mixture or shunt like, the value of which is on the upper border line. As HR is abnormally high, the elevated A-a PO2 was attributed to VA/Q mismatch and that was mainly induced by abnormally rapid pulmonary circulation. Oxygen consumption showed normal values but not optimal amount relative to abnormally high oxygen delivery, the product of cardiac output and arterial content. It is very important to note that VD/VT ratio was elevated due to high cardiac output that contributed to VA/Q mismatch. Beside VA/Q mismatch and the elevated VD/VT, the SaO2 of 91% approached the low boundary.
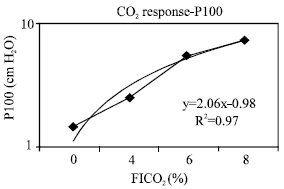 | |
| Fig. 1: | P100 during CO2 response study |
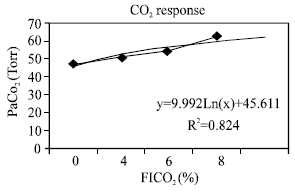 | |
| Fig. 2: | Arterial PCO2 during CO2 response study |
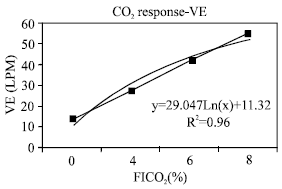 | |
| Fig. 3: | Ventilation during CO2 response |
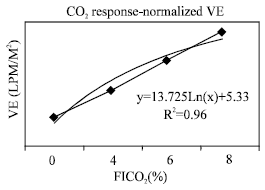 | |
| Fig. 4: | Normalized ventilation during CO2 response |
Thus the group was classified into moderate hypoxemia during awake, with potential for further de saturation.
| Table 1: | Anthropometric and pulmonary functions of patients |
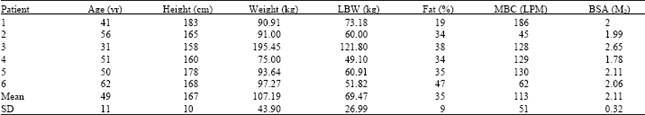 | |
| Table 2: | Sleep study summary report that contains apnea indices and oxygen saturation during sleep |
 | |
| *Obstructive index: Chest and abdominal effort present with flow absence **Central index: Chest and abdominal effort and flow are absent. ***Mixed index: Initiated as central then followed by obstructive | |
| Table 3: | Oxygenation and gas exchange variables |
| Measurements were made in triplicate and average was recorded for each patient.* Data in the table are the average of the six patients±SD is reported in the table | |
| Table 4: | Steady-state hypercapnea-induced hypercapnea study |
 | |
| *Measurements were made in triplicate and average was recorded for each patient Data in the table are the average of the six patients±SD | |
Evaluation of hypercapnea response: The transient pressure generation, P100 (Fig. 1) which was used as an approximation for diaphragm activation increased linearly (R2=0.96) and in proportion to increasing CO2 concentration in inspired air from 0-8%. Thus the overall response means that the diaphragm was receiving sufficient stimuli in response to hypercapnea stimulus. As the majority of the stimulus is mediated by phrenic nerve, the possibility of impairment of the respiratory center was ruled out. In addition, the partial pressure of arterial CO2 (PaCO2) was elevated at room air, indicating lower alveolar ventilation and higher CO2 stores. Moreover, physiologic dead space (VD/VT) ratio was abnormal as compared with normal values at all levels of PaCO2 and all levels of FICO2 (Fig. 2). It is clear that hypoventilatory response in this group of patients was impaired via airway obstruction rather than central control related mechanisms. Figure 3 showed that maximum ventilation (VEmax), on FICO2 of 8% was subnormal as compared to average population values of 150 LPM. Both VEmax and normalized VEmax were subnormal in comparison with normal population values (Table 4). The VE regression lines from the onset of the experiments to VEmax are illustrated in Fig. 3 and 4. The regression models of VE on FICO2 from 0-8% are displayed on both figures. The model showed a reduction in VE/FICO2 ratio as FICO2 increased which implied respiratory muscle fatigue as maximal VE was approached.
DISCUSSION
There are two major findings from this study; OSA have an elevated cardiac output during daytime which was related to high blood pressure; and hypoventilatory response to hypercapnea which was associated with high transient pressure generation (P100).
The linear rise in P100 without proportion increase in VE, reflected an elevated intrathoracic pressure that characterize obstructive sleep apnea during wake state. As cardiac output was elevated, these findings does support the concept that rely on changes in intrathoracic pressure could alter filling of the right and left heart through its effect on venous return and intraventricular dependence[20-22]. It can also support the concept that rely on changes in intrathoracic pressure affect afferent neural traffic from the chest wall and lung receptors[23-27], as evident by elevated heart rate and tension time index. Present results reproduce real life important characteristics of obstructive sleep apnea, namely, sleep-related hypoxemia and sleep fragmentation as evident from 24 h sleep study. Together, it can be stated that the acute and chronic hypertension caused by OSA can in turn exacerbate OSA as the two diseases may be linked via positive feedback rather than negative feed forward mechanisms.
Several authors have reported a remarkably high prevalence of OSA in hypertensive compared to normotensive patients[6,28]. Other studies have demonstrated that the prevalence of hypertension among patients with OSA is higher than in the general population[29-31]. A major problem with this type of epidemiological evidence is the presence of confounding variables, particularly obesity[32,33] that predispose to both OSA and hypertension. Nevertheless, even in studies in which obesity, gender and age were statistically controlled, sleep apnea continued to be an independent risk factor for hypertension. A few clinical studies have described a decrease in blood pressure after effective treatment of OSA[34,35], but interpretation of these studies is complicated by concurrent changes in body mass, alcohol consumption and antihypertensive medications as well as the direct effects of treatment on the cardiovascular system, such as continuous positive airway pressure.
In contrast to these epidemiological and clinical studies, the relationship between cardiac output and OSA found in the present study demonstrated a direct link between OSA and hypertension in the absence of confounding variables. Present findings are likely to be relevant to OSA in humans. Moreover, the results of our study indicated that disruption of sleep pattern by recurrent arousals is not the underlying stimulus, suggesting that less sensitivity response to hypercapnea which could be also related to fluctuations in intrathoracic pressure.
In conclusion, present study used real life human OSA patients in whom recurrent upper airway occlusion during sleep but lower hypercapnea responses associated with high pressure generation and elevated cardiac output during awake. These findings suggest that OSA is a risk factor for heart disease if hypercapnea-induced-hyperapnea test results did not rule out hypoventilatory response during wake state. In addition, the modified steady state hypercapnea test is simple, cost effective and can be done noninvasive to rule out risk for heart diseases in OSA.
REFERENCES
- Haffor, A.S.A. and J.G. Nail, 1988. Microprocessor-controlled gas mixing system for rebreathing equilibration. J. Comput. Biomed. Res., 21: 101-109.
PubMed - Fung, J.W., T. Li, D.K. Choy, G. Yip, F.S. Ko, J.E. Sanderson and D.S.C. Hui, 2002. Severe obstructive sleep apnea is associated with left ventricular diastolic dysfunction. Chest, 21: 422-429.
CrossRef - Kales, A., R.J. Cadieux, L.C. Shaw, A. Vela-Bueno and E.O. Bixler et al., 1984. Sleep apnoea in a hypertensive population. Lancet, 324: 1005-1008.
CrossRef - Hla, K.M., T.B. Young, T. Bidwell, M. Palta, J.B. Skatrud and J. Dempsey, 1994. Sleep apnea and hypertension: A population-based study. Ann. Inter. Med., 120: 382-388.
CrossRefDirect Link - Fogel, R.B., A. Malhotra, G. Pillar, S.D. Pittman, A. Dunaif and D.P. White, 2001. Increased prevalence of obstructive sleep apnea syndrome in obese women with polycystic ovary syndrome. J. Clin. Endocrinol. Metabol., 86: 1175-1180.
CrossRef - Motta, H., C. Guilleminault, J.S. Schroeder and W.C. Dement, 1978. Tracheotomy and homodynamic changes in sleep-induced apnea. Ann. Int. Med., 89: 454-458.
PubMed - Haffor, A.S.A., R.L. Bartels, R.L. Hamlin, T.E. Kirby and A.L. Kunze, 1987. Carbon dioxide storage capacity of endurance and sprint-trained athletes in exercise. Arch Int Physiol Biochim., 95: 81-90.
PubMed - Fletcher, E.C., J. Lesske, W. Qian, C.C. Miller and T. Unger, 1992. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension, 19: 555-561.
Direct Link - Bender, P.R., B. Groves, R.E. McCullough, R.G. McCullough and S.Y. Hoang et al., 1998. Oxygen transport to exercising leg in chronic hypoxia. J. Applied Physiol., 65: 2592-2597.
PubMed - Ringler, J., B.C. Basner, R. Shannon, R. Schwartzstein, H. Manning, S.E. Weinberger and J.W. Weis, 1990. Hypoxemia alone does not explain blood pressure elevations after obstructive apneas. J. Applied Physiol., 69: 2143-2148.
PubMed - Brooks, D., R.L. Horner, L.F. Kozar, T.K. Waddell, C.L. Render and E.A. Phillipson, 1996. Validation of a telemetry system for long-term measurement of blood pressure. J. Applied Physiol., 81: 1012-1018.
PubMed - Phillipson, E.A., E. Murphy and L.F. Kozar, 1976. Regulation of respiration in sleeping dogs. J. Applied Physiol., 40: 688-693.
PubMed - Robotham, J.L., J. Rabson, S. Permutt and B. Bromberger-Barnea, 1979. Left ventricular hemodynamics during respiration. J. Applied Physiol., 47: 1295-1303.
PubMed - Scharf, S.M., R. Brown, N. Saunders and L.H. Green, 1979. Effects of normal and loaded spontaneous inspiration on cardiovascular function. J. Applied Physiol., 47: 582-590.
PubMed - Tarasiuk, A. and S.M. Scharf, 1993. Effects of periodic obstructive apneas on venous return in closed-chest dogs. Am. Rev. Respir. Dis., 148: 323-329.
Direct Link - Davenport, P.W., F.J. Thompson, R.L. Reep and A.N. Freed, 1985. Projection of phrenic nerve afferents to the cat sensorimotor cortex. Brain Res., 328: 150-153.
CrossRef - Issa, F.G., S.G. McNamara and C.E. Sullivan, 1987. Arousal responses to airway occlusion in sleeping dogs: Comparison of nasal and tracheal occlusions. J. Applied Physiol., 62: 1832-1836.
PubMed - Gandevia, S.C. and G. Macefield, 1989. Projection of low-threshold afferents from human intercostal muscles to the cerebral cortex. Respir. Physiol., 77: 203-214.
PubMed - Gleeson, K., C.W. Zwillich and D.P. White, 1990. The influence of increasing ventilatory effort on arousal from sleep. Am. Rev. Respir. Dis., 142: 295-300.
PubMed - Kimoff, R.J., H. Makino, R. Horner, L.F. Kozar, F. Lue, A.S. Slutsky and E.A. Phillipson, 1994. Canine model of obstructive sleep apnea: Model description and preliminary application. J. Applied Physiol., 76: 1810-1817.
PubMed - O'Donnell, C.P., E.D. King, A.R. Schwartz, J.L. Robotham and P.L. Smith, 1994. Relationship between blood pressure and airway obstruction during sleep in the dog. J. Applied Physiol., 77: 1819-1828.
PubMed - He, J., M.H. Kryger, F.J. Zorick, W. Conway and T. Roth, 1988. Mortality and apnea index in obstructive sleep apnea. Experience in 385 male patients. Chest, 1: 9-14.
PubMed - Williams, A.J., D. Houston, S. Finberg, C. Lam, J.L. Kinney and S. Santiago, 1985. Sleep apnea syndrome and essential hypertension. Am. J. Cardiol., 55: 1019-1022.
CrossRef - Hirshkowitz, M., I. Karacan, A. Gurakar and R.L. Williams, 1989. Hypertension, erectile dysfunction and occult sleep apnea. Sleep, 12: 223-232.
PubMed - Chin, K., K. Shimizu, T. Nakamura, N. Narai and H. Masuzaki et al., 1999. Changes in intra-abdominal visceral fat and serum leptin levels in patients with obstructive sleep apnea syndrome following nasal continuous positive airway pressure therapy. Circulation, 100: 706-712.
Direct Link - O'Donnell, C.P., C.G. Tankersley, V.P. Polotsky, A.R. Schwartz and P.L. Smith, 2000. Leptin, obesity and respiratory function. Respir. Physiol., 119: 163-170.
CrossRef - Burack, B., 1984. The hypersomnia-sleep apnea syndrome: Its recognition in clinical cardiology. Am. Heart J., 107: 543-548.
Direct Link - Grunstein, R., I. Wilcox, T.S. Yang, Y. Gould and J. Hedner, 1993. Snoring and sleep apnoea in men: Association with central obesity and hypertension. Int. J. Obesity, 17: 533-540.
PubMed