
Mohammad A. Khaled
Department of Nutrition
Sciences,
University of Alabama at
Birmingham Medical Center,
Birmingham, Alabama 35294,
USA
Journal of Medical Sciences
Year: 2002 | Volume: 2 | Issue: 1 | Page No.: 44-46







ABSTRACT
Traditional cardiovascular risk factor, such as hyper cholesterolemia, smoking, diabetes, and hypertension, do not necessarily explain the incidence of heart disease. Several less substantiate risk factors may contribute significantly to cardiovascular Pathogenesis. H. pylori is a gram-negative, microaerophilic, spiral bacterium that occurs naturally, and inhabits the mucus layer that covers the gastric epithelial cells. This Short review deals with the heart disease due to infection, H. pylori.
PDF Abstract XML References Citation
How to cite this article
Mohammad A. Khaled, 2002. Heart Disease Due to Infections: the Helicobacter pylori. Journal of Medical Sciences, 2: 44-46.
DOI: 10.3923/jms.2002.44.46
URL: https://scialert.net/abstract/?doi=jms.2002.44.46
DOI: 10.3923/jms.2002.44.46
URL: https://scialert.net/abstract/?doi=jms.2002.44.46
In recent years, observational studies in humans have provided intriguing hints linking certain viral and bacterial infections to coronary heart disease (CHD) (Gura, 1998; Libby et al., 1997; Lip and Beevers, 1997). Mechanistic studies suggested that gene products of infectious agents may acts directly on the vascular cells. Nevertheless, an important question is, whether infectious agents can exert vasculopathic effects without infecting vascular cells. For example, the atherogenic changes seen in infected endothelial and smooth muscle cells can be caused by systemic inflammation due to chronic infection and activation of circulating inflammatory cells. It is also possible that infectious agents currently showing weak associations with vascular disease, as well as with infectious agents which were not previously associated with atherogenesis, may contribute to vascular pathogenesis. Rupprecht et al. (2001) analyzed infectious burden with 8 pathogens in 1018 CHD patients and found that only Epstein-Barr virus, Helicobacters pylori and herpes simplex virus type 2 were independently associated with the potential death from CHD.
H. pylori originally thought to be a member of the genus Campylobacter, occurs naturally and inhabits the mucus layer that covers the gastric epithelial cells (Dekigai et al., 1995). It is a gram-negative, microaerophilic, spiral bacterium that possesses a powerful enzyme, urease. Urease protects this organism and allows it to survive in an acidic environment by producing ammonia that increases the pH in immediate vicinity (Booth, 1985). The ammonia gas is toxic to gastric and other epithelial cells resulting in hypochlorhydria in the stomach (Suzuki et al., 1992). H. pylori is commonly accepted as the etiological agent of type B gastritis and as a major contributor to peptic ulcer disease (Graham, 1989). Several specific virulence factors such as cytotoxin-associated gene A (cagA) and vacuolating cytotoxin A (vacA) as well as other noxious substances including ammonia, lipopolysaccharide (endotoxin), platelet activating factor (PAF), nitric oxide (NO), have been implicated in gastritis (Perri et al., 1999; Cover and Blaser, 1996). Chronic inflammation, atrophic gastritis, intestinal metaplasia, impaired micronutrient absorption (ascorbic acid and vitamin B12 in particular) combined with hypergastrinemia in the stomach, excessive reactive oxygen species (ROS) and epithelial cell proliferation have all been associated with gastric cancer due to H. pylori (Blaser, 1992; Correa, 1995; Konturek et al., 1999). Consequently we found a state of oxidative stress, as defined by an excessive pro-oxidant, i.e., ROS, activity with concomitant inadequate/compromised antioxidant protection, in H. pylori infected human subjects (Khaled and Sarker, 1998). Oxidative state, at least in terms of lipid per oxidation, plays a greater role in the pathogenesis of heart disease (Khan and Baseer, 2000).
Sung and Sanderson (1996) and Markle (1997) hypothesized possible mechanisms of association of H. pylori infection and CHD involving hyperhomocysteinemia which is caused by the over-production of total homocysteine (tHCY) (Goraham, 1997). Basic etiological factors for homocysteine formation are the following:
| a) | Nutritional deficiencies of folic acid, vitamin B12 and/or vitamin B6. |
| b) | There could be a faulty transport protein for any of the above vitamins/cofactors. |
| c) | There may be a genetic component. For example, the genetically heterozygous loss of cystathionine β-synthase in the transsulfuration pathway results in the disease homocystinuria. This disease causes profound cardiovascular disease. Another genetic disease affecting tHCY levels in plasma is a mutation of the methylene tetrahydrofolate reductase (MTHFR) gene, which results in a thermolabile enzyme, the end result of which is a large reduction of methyl groups being made available to the remethylation pathway, and the subsequent build-up of homocysteine. |
Soon after Sung and Sanderson (1996) proposed their hypothesis, a handful of investigators claimed that no such association of tHCY could be found in CHD patients infected with H. pylori (Whincup et al., 1997; Saxena et al., 1997; Leung et al., 2001). In the absence of many other pieces of information, particularly patients' dietary habits, Menge et al. (2000) commented that the proposed link between tHCY and H. pylori needed further investigations. For example, if the patients with and without H. pylori infection did have adequate and/or equal intake of the above mentioned nutrients, these observations might have been confounded. Additionally, Chambers et al. (2000) reported tHCY-induced abnormal endothelial function in humans due to the deficiency of the above mentioned B vitamins. Interestingly, such adverse effects due to tHCY were reversed by supplementation with vitamin C (Chambers et al., 1999). We and others have shown that H. pylori is capable of depleting these micro nutrients in humans (Carmel et al., 1994; Khaled et al., 1997).
Lip et al. (1996) reported an important finding that the body mass index (BMI), an approximate measure of body fatness, was significantly higher in H. pylori seropositive than seronegative patients with CHD; (BMI 28.4 ± 4.2 vs 26.2 ± 3.2 p = 0.019). Mendall et al. (1997) also reported a strong association between BMI and TNF-alfa in H. pylori infected CHD patients. Recently, we, found a significant difference in TNF-alfa and TBARS (thiobarbitoric acid reacting substance, as an index of lipid per oxidation) between H. pylori positive and negative healthy human subjects with similar BMI (Khaled and Mahalanabis, 2000). Interestingly, H. pylori infection has also been found to modify serum lipid, particularly triglyceride (TG), concentrations (Laurila et al., 1999) and higher serum TG is associated with higher lipid per oxidation, a known risk factor for CHD (Khan and Baseer, 2000). Recently, it has been well documented that the level of lipid per oxidation increased linearly with increased levels of tHCY (Welch and Loscalzo, 1998).
After having described our and others studies on H. pylori, related to its associations with heart disease, probable mechanisms of H. pylori actions could be visualized by mapping its pathways as depicted in the diagram below.
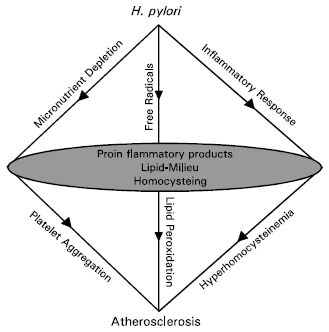 |
The circulating lipid-milieu in this diagram is a major host factor. This host factor could be exacerbated either by the host’s visceral fat/TG and/or by H. pylori infection and/or by the host’s dietary habits. Per oxidation of this lipid-milieu, leading to atherosclerosis, could be induced by some inflammatory/ proinflammatory products as generated by H. pylori. Hyperhomocysteinemia, as a consequence of H. pylori infection, is also capable of inducing atherosclerosis either by per oxidation of the lipid milieu and/or by promoting platelet aggregation. Depletion of antioxidant protection, as created by this bacterium, could very well facilitate development and/or aggravate incidence of atherosclerosis in humans. Prospective scientific investigations in this direction will be necessary to substantiate this model of H. pylori actions associated with the induction and/or progression of CHD in humans.
REFERENCES
- Carmel, R., G.I. Perez-Perez and M.J. Blaser, 1994. Helicobacter pylori infection and food-cobalamin malabsorption. Digestive Dis. Sci., 39: 309-314.
CrossRefDirect Link - Correa, P., 1995. The role of antioxidants in gastric carcinogenesis. Crit. Rev. Food Sci. Nutr., 35: 59-64.
CrossRefPubMedDirect Link - Cover, T.L. and M.J. Blaser, 1996. Helicobacter pylori infection, a paradigm for chronic mucosal inflammation: Pathogenesis and implications for eradication and prevention. Adv. Interal Med., 41: 85-117.
PubMedDirect Link - Dekigai, H., M. Murakami and T. Kita, 1995. Mechanism of Helicobacter pylori-associated gastric mucosal injury. Digestive Dis. Sci., 40: 1332-1339.
CrossRefDirect Link - Goraham, I.M., L.E. Daly, H.M. Refsum, K. Robinson and L.E. Brattstrom et al., 1997. Plasma homocysteine as a risk factor for vascular disease: The European concerted action project. J. Am. Med. Assoc., 277: 1775-1781.
CrossRefDirect Link - Graham, D.Y., 1989. Campylobacter pylori and peptic ulcer disease. Gastroenterology, 5: 615-625.
CrossRefDirect Link - Gura, T., 1998. Infections: A cause of artery-clogging plaques? Science, 281: 35-37.
CrossRefDirect Link - Sarker, S.A., M.A. Wahed and M.A. Khaled, 1997. H. pylori infection and vitamin a depletion in malnourished children. Gastroenterology, 112(4 Suppl.): A278-A278.
Direct Link - Khaled, M.A. and S.A. Sarker, 1998. Changes of oxidant and antioxidant status in humans due to H. Pylori infection. Nutr. Res., 18: 1463-1468.
CrossRefDirect Link - Konturek, P.C., W. Bielanski, S.J. Konturek and E.G. Hahn, 1999. Helicobacter pylori associated gastric pathology. J. Physiol. Pharmacol., 50: 695-710.
PubMedDirect Link - Laurila, A., A. Bloigu, S. Nayha, J. Hassi, M. Leinonen and P. Saikku, 1999. Association of Helicobacter pylori infection with elevated serum lipids. Atherosclerosis, 142: 207-210.
CrossRefPubMedDirect Link - Leung, W.K, P.K. Ma, P.C.L. Choi, J Y.L. Ching and A.C.W. Ng et al., 2001. Correlation between Helicobacter pylori infection, gastric inflammation and serum homocysteine concentration. Helicobacter, 6: 146-150.
Direct Link - Libby, P., D. Egan and S. Skarlatos, 1997. Roles of infectious agents in atherosclerosis and restenosis: An assessment of the evidence and need for future research. Circulation, 96: 4095-4103.
PubMedDirect Link - Lip, G.Y.H., R. Wise and G. Beevers, 1996. Association of Helicobacter pylori infection with coronary heart disease. Study shows association between H. pylori infection and hypertension. Br. Med. J., 312: 250-251.
PubMedDirect Link - Lip, G.Y.H. and D.G. Beevers, 1997. Can we treat coronary artery disease with antibiotics? Lancet, 350: 378-379.
CrossRefPubMedDirect Link - Markle, H.V., 1997. Coronary artery disease associated with Helicobacter pylori infection is at least partially due to inadequate folate status. Med. Hypotheses, 49: 289-292.
CrossRefDirect Link - Menge, H., A. Lang, B. Brosius, R. Hopert and H. Lollgen, 2000. Helicobacter pylori and coronary heart diseases-hypotheses and facts. Z. Gastroenterol., 38: 315-323, (In German).
CrossRefPubMedDirect Link - Khan, M.A. and A. Baseer, 2000. Increased malondialdehyde levels in coronary heart disease. J. Pak. Med. Assoc., 50: 261-264.
Direct Link - Perri, F., R. Clemente, V. Festa, C.C. de Ambrosio and M. Quitadamo et al., 1999. Serum tumour necrosis factor-alpha is increased in patients with Helicobacter pylori infection and CagA antibodies. Ital. J. Gastroenterol. Hepatol., 31: 290-294.
PubMedDirect Link - Rupprecht, H.J., S. Blankenberg, C. Bickel, G. Rippin and G. Hafner et al., 2001. Impact of viral and bacterial infectious burden on long-term prognosis in patients with coronary artery disease. Circulation, 104: 25-31.
CrossRefPubMedDirect Link - Saxena, V., H. Markus, S. Swaminathan and M.E. Mendall, 1997. Hyperhomocysteinaemia, Helicobacter pylori and coronary heart disease. Heart, 78: 524-524.
PubMedDirect Link - Sung, J.J. and J.E. Sanderson, 1996. Hyperhomocysteinaemia, Helicobacter pylori and coronary heart disease. Heart, 76: 305-307.
CrossRefPubMedDirect Link - Suzuki, M., S. Miura, M. Suematsu, D. Fukumura and I. Kurose et al., 1992. Helicobacter pylori-associated ammonia production enhances neutrophil-dependent gastric mucosal cell injury. Am. J. Physiol., 263: G719-G725.
PubMedDirect Link - Chambers, J.C., A. McGregor, J. Jean-Marie, O.A. Obeid and J.S. Kooner, 1999. Demonstration of rapid onset vascular endothelial dysfunction after hyperhomocysteinemia: An effect reversible with vitamin C therapy. Circulation, 99: 1156-1160.
PubMedDirect Link - Chambers, J.C., O.A. Obeid, H. Refsum, P. Ueland and D. Hackett et al., 2000. Plasma homocysteine concentrations and risk of coronary heart disease in UK Indian Asian and European men. Lancet, 355: 523-527.
CrossRefPubMedDirect Link - Blaser, M.J., 1992. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology, 102: 720-727.
CrossRefPubMedDirect Link - Booth, I.R., 1995. Regulation of cytoplasmic pH in bacteria. Microbiol. Rev., 49: 359-378.
PubMedDirect Link - Mendall, M.A., P. Patel, M. Asante, L. Ballam and J. Morris et al., 1997. Relation of serum cytokine concentrations to cardiovascular risk factors and coronary heart disease. Heart, 78: 273-277.
CrossRefDirect Link - Welch, G.N. and J. Loscalzo, 1998. Homocysteine and atherothrombosis. N. Engl. J. Med., 338: 1042-1050.
CrossRefPubMedDirect Link