
Jessica Varela Chaves
Universidade do Estado do Amazonas (UEA), Escola Superior de Ciencias da Saude, Pos-Graduacao em Biotecnologia E Recursos Naturais, Manaus, Amazonas, Brasil
Cinthya Paola Ortiz Ojeda
Universidade do Estado do Amazonas (UEA), Escola Superior de Ciencias da Saude, Pos-Graduacao em Biotecnologia E Recursos Naturais, Manaus, Amazonas, Brasil
Ingrid Reis da Silva
Universidade Federal do Amazonas (UFAM)-Programa Multi-Institucional de Pos-Graduacao em Biotecnologia, Manaus, Amazonas, Brasil
Rudi Emerson de Lima Procopio
Universidade do Estado do Amazonas (UEA), Escola Superior de Ciencias da Saude, Pos-Graduacao em Biotecnologia E Recursos Naturais, Manaus, Amazonas, Brasil
LiveDNA: 55.16290
Research Journal of Microbiology
Year: 2018 | Volume: 13 | Issue: 1 | Page No.: 13-20







ABSTRACT
Background and Objective: A comprehensive, phylogeny of genus Streptomyces is needed for a better understanding of their ecology as well as for facilitating their bioprospecting. 16S-rRNA-based phylogenetic reconstruction does not guarantee well-resolved and robust trees that reflect the overall relationship between Streptomyces species, therefore it is necessary to find a region of the genome that best shows the difference between Streptomyces. The goal of the present study was to produce a more robust phylogeny for Streptomyces by comparing the phylogenetic trees derived from concatenated gene and single gene sequence data. Methodology: Improvements in DNA sequencing technologies have resulted in the ability to generate large numbers of high quality draft genomes that have led to a dramatic increase in the number of publically available genomes and this has allowed researchers to characterize microorganisms using genomic data. In the present study, a phylogeny of 26 Streptomyces strains were analyzed using individual genes with more than 1 kb and compared with a phylogeny of 8 highly informative concatenated genes, for a total of 20 kb. Analyses were performed in MEGA, which defined the topology of the consensus tree. Results: The results from the concatenated genes showed a much higher power of discrimination and a much more stable topological structure than the 16S rRNA gene, with clearly better discriminated entities and higher bootstrap support. Comparing the 23S rRNA gene tree with the concatenated gene tree, it was found that the 23S rRNA tree had discriminatory power and topological stability similar to the concatenated gene tree. Conclusion: It is concluded that the 23S gene can be used as an alternative to 16S for the identification and classification of streptomycetes at species and intraspecies levels. The inner fragment of 23S (from 1 to 2 kb) is the most variable region and generated reliable and robust trees.
PDF Abstract XML References Citation
Received: May 05, 2017;
Accepted: July 21, 2017;
Published: December 15, 2017
How to cite this article
Jessica Varela Chaves, Cinthya Paola Ortiz Ojeda, Ingrid Reis da Silva and Rudi Emerson de Lima Procopio, 2018. Identification and Phylogeny of Streptomyces Based on Gene Sequences. Research Journal of Microbiology, 13: 13-20.
URL: https://scialert.net/abstract/?doi=jm.2018.13.20
URL: https://scialert.net/abstract/?doi=jm.2018.13.20
INTRODUCTION
Streptomyces species are among the best studied and characterize organisms, because of their importance in the production of substances with medical applications1. The importance of streptomycetes to medicine results from their known ability to produce over two-thirds of the naturally derived antibiotics in current use (and many other pharmaceuticals such as anti-tumour agents and immunosuppressants) through means of complex secondary metabolic pathways2. Therefore, Streptomyces is one of the most important sources of bio-active molecules for medicine and industry3. There have been efforts to establish a comprehensive, detailed and robust phylogeny of Streptomyces based on single gene and genomic4. Currently available phylogenies of the group are based on the 16S rRNA gene; however, such reconstructions tend to be relatively unstable and are not guaranteed to reflect the overall evolutionary history in a complex group, with widespread horizontal gene transfer, such as Streptomyces4. Despite the 16S rRNA sequences from almost all Streptomyces type strains being available in public databases, contributed by researchers from several countries and the phylogenies being presented in the literature, many species relationships within Streptomyces remain unclear. Streptomyces has become one of the most taxonomically complex groups5, with the majority of its species sharing highly similar phenotypes and 16S rRNA sequences6,7. The use of whole genome sequences has been regarded as a promising avenue for the future of Streptomyces taxonomic and phylogenetic studies. Since rapid improvements in DNA sequencing technologies are providing new approaches to address major questions in the field of microbial taxonomy8-10. The goal of the present study was to produce a more robust phylogeny for Streptomyces by comparing the phylogenetic trees derived from concatenated gene and single gene sequence data.
MATERIALS AND METHODS
This study was conducted at the University of the State of Amazonas, UEA, in the postgraduate Biotechnology Laboratory, 2016.
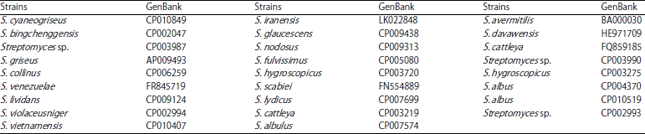
Genomes used: The National Center for Biotechnology Information (NCBI) is well known for the nucleotide sequence archive, GenBank and sequence analysis tool BLAST. A total of 26 Streptomyces genomes from different species and strains of interest were randomly retrieved from NCBI and used in this study, the lineages and the number in GenBank are listed in the Table 1.
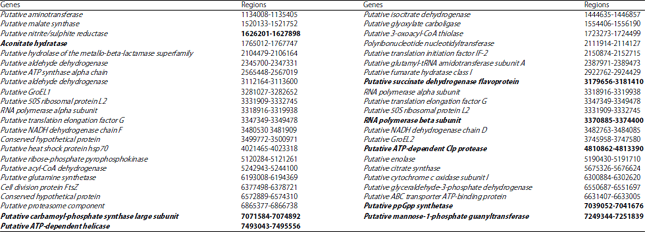
The similarity to Streptomyces griseus NBRC 13350 (AP009493.1) was evaluated through BLASTn (Basic Local Alignment Search Tool), with genes being selected from the strains based on size higher than 1 kb and high similarity. The genes that exhibit a greater similarity are listed in Table 2. Sequence Manipulation Suite (Reverse Complement)11 was used to invert the genomes that were reversed.
Phylogenetic analysis: One goal of study was to reevaluate the phylogenetic relationships of Streptomyces species by using different genes (Table 2) with the concatenated gene tree and to compare between the trees generated by the 16S and 23S rRNA genes. Sequences were aligned using CLUSTAL_W. To ensure the stability and reliability of phylogenetic relationships among strains used in this study, phylogenetic trees were constructed through the neighbour-joining (NJ) method. Analyses were performed in MEGA 5.212 and bootstrap13 was calculated to determine branch support (from 500 resembling). Analyses were performed for each of the 40 genes (Table 2) so they could be compared with the tree from the concatenated gene analysis.
Concatenated gene analysis: For the concatenated gene phylogenetic analysis, 8 conserved genes sequences (Table 2, highlighted in bold) were individually aligned using Clustal Omega14 and after removing all sites containing gaps, the 8 sequences were concatenated manually into one meta-alignment.
| Table 1: | Strains of Streptomyces used in this study and GenBank accession numbers |
 | |
| Table 2: | Genes of S. griseus NBRC 13350 with greater similarity between the strains of Streptomyces and region in the genome of NBRC 13350 |
 | |
The meta-alignment contained a total of 20,000 phylogenetically informative sites. Bootstrap was calculated for branch support (500 resembling).
RESULTS
After analysis of the genes with more than 1 kb of S. griseus NBRC 13350, it was observed that the aconitate hydratase, putative succinate dehydrogenase flavoprotein, RNA polymerase beta subunit, putative ATP-dependent Clp protease, putative ppGpp synthetase, putative carbamoyl-phosphate synthase large subunit, putative mannose-1-phosphate guanyltransferase, putative ATP-dependent helicase and 23S genes, presented better similarity and higher value of bootstrap and better distribution of the species in the phylogenetic tree. All gene trees were compared with the tree of the 8 concatenated genes, with 23S being the one that showed the best similarity with the concatenated gene tree.
The alignment of the 16S rRNA gene sequences showed a high similarity between strains as indicated by the scale bar in Fig. 1. There was poor topological congruence between the 16S tree and the other trees. Among all trees, bootstrap supports were the lowest in the 16S tree. Overall, the results showed that the strains are very closely related (Fig. 1).
The phylogenetic tree based on the eight concatenated genes had a different topology than the 16S rRNA gene tree and most of the 16S rRNA gene clusters were not recovered. The results from the concatenated genes showed a much higher power of discrimination and a much more stable topological structure than the 16S rRNA gene, with clearly better discriminated entities and higher bootstrap support (Fig. 2).
Comparing the 23S rRNA gene tree (Fig. 3) with the concatenated gene tree, it was found that the 23S rRNA tree had discriminatory power and topological stability similar to the concatenated gene tree. It was also found that the 23S rRNA gene tree had good resolution and robustness. Although a few branches were poorly resolved or showed dissimilar structures when compared to the concatenated gene tree, it was still efficient in differentiating most of the strains (Fig. 3).
The ideal means of identifying and classifying bacteria would be to compare each genome in a given strain with the genome of all known species. This cannot be done but the gene of one organism can be compared with that of any other organism.
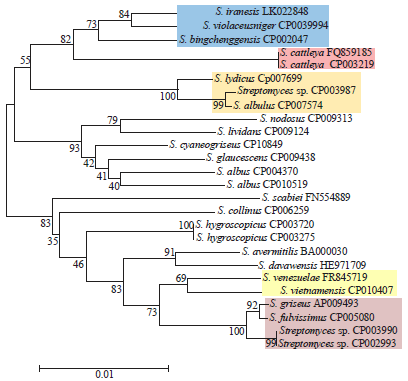 | |
| Fig. 1: | Phylogenetic relationships among 26 Streptomyces strains based on 16S rRNA gene sequences |
The tree was constructed using the NJ method. Numbers at nodes represent levels (%) of bootstrap support from 500 resampled datasets. The bar indicates 1% estimated sequence divergence. Strains of clusters I, II and III are highlighted | |
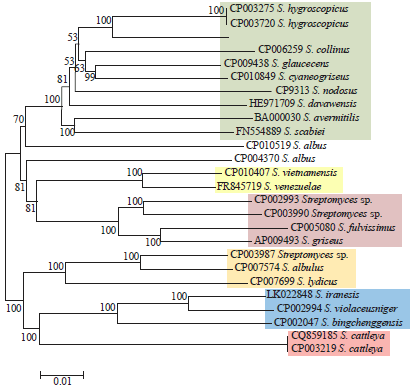 | |
| Fig. 2: | Phylogenetic relationships among 26 Streptomyces strains based on eight concatenated gene sequences |
The tree was constructed using the NJ method. Numbers at nodes represent levels (%) of bootstrap support from 500 resampled datasets. The bar indicates 1% estimated sequence divergence | |
The 23S gene is around 3 kb, thus it was chose a partial and less conserved region, an inner fragment of the gene (between nucleotide 1000 and 2000), which gave enough resolution to separate closely related Streptomyces species.
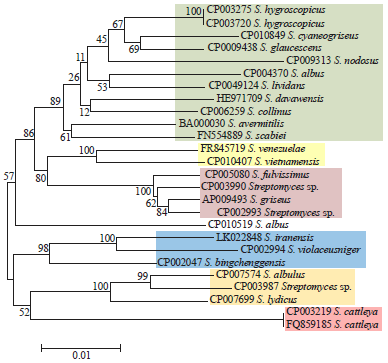 | |
| Fig. 3: | Phylogenetic relationships among 26 Streptomyces strains based on 23S rRNA gene sequences |
The tree was constructed using the NJ method. Numbers at nodes represent levels (%) of bootstrap support from 500 resampled datasets. The bar indicates 1% estimated sequence divergence | |
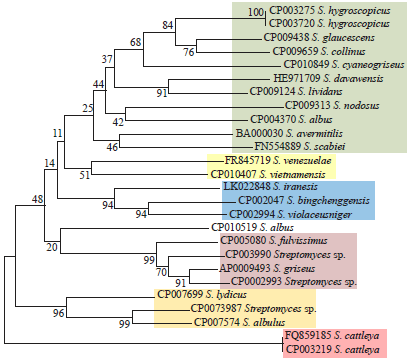 | |
| Fig. 4: | Phylogenetic relationships among 26 Streptomyces strains based on partial 23S rRNA gene sequences |
The tree was constructed using the NJ method. Numbers at nodes represent levels (%) of bootstrap support from 500 resampled datasets. The bar indicates 1% estimated sequence divergence | |
The 23S inner region tree usually had higher bootstrap values than those of the complete 23S dataset. Topologies of both trees of 23S RNA gene were very similar, but the estimated sequence divergences were different as can be seen in the bars of Fig. 3 and 4, the latter being shorter, showing a larger difference between species.
The partial 23S rRNA tree proved to be the most robust and viable phylogenetic tree that differentiated most strains in this study, with an identification sequence less than 1000 bp long, to use only one reaction in Sanger sequencing technology.
Understanding the extent of genetic and functional diversity among strains of the same or very closely related, species has become a cornerstone issue for bacterial systematics, especially for Streptomyces, which produces important secondary metabolites15. Analysis using 16S rRNA genes is frequently used to obtain the taxonomic composition of a microbial community16, 17.
DISCUSSION
Several phylogenetic trees were constructed, using all the genes of Table 2 plus the 16S and 23S and compared these trees with the 8 concatenated genes. A variation in the bar scale was observed, showing the genetic change of the genus Streptomyces. With more conserved genes and genes with higher numbers of changes but the distribution of the species in the phylogenetic tree does not suffer many changes from one gene to another.
Genes used in molecular systematics should be evaluated for their phylogenetic performance from previous studies18. Protein-coding genes aren’t commonly used in the identification of bacteria and some of them have been used individually for phylogenetic analyses of Streptomyces4,19 demonstrating that they can give a higher resolution for the phylogeny of Streptomyces. The 16S rRNA gene tree is unreliable due to the conflicting topologies obtained and the low bootstrap support values observed, which might indicate an incorrect relationship between the Streptomyces strains studied. Guo et al.6 stated that the 16S rRNA gene is more appropriate for the discrimination of distantly related streptomycetes, but is not efficient for closely related strains.
The 16S rRNA gene sequence has been determined for a large number of Streptomyces strains. GenBank, the largest databank of nucleotide sequences has millions of deposited sequences and accepts any linked name and sequence that is sent to it. Often, the sequences deposited in GenBank are not complete or as accurate as they should be. Thus, there are many deposited sequences that are comparable to an unknown strain, indicating that many genetically different strains were being deposited under the same species name. For phenotypic identification of micro-organisms, it depended on a database with accurate morphological and biochemical descriptions of typeor typical, strains and of standard methods to determine these characteristics in the isolate to being identified20. Similarly, for accurate organism identification through 23S rRNA gene sequences, it is important to databases with accurately identified sequences and a high quality sequence from the isolate to be identified. The 23S sequences deposited in GenBank are of better quality than the 16S ones, because most are from Streptomyces standard strains and some have their whole genome described, a better gene for comparison overall.
Streptomyces is an important group for industrial microbiology, however, its species are usually difficult to identify1 and the correct designation of Streptomyces strains is important to better discriminate between them. The 16S rRNA gene sequences provide limited identification of Streptomyces strains. An additional important function of 23S rRNA gene sequencing is to provide accurately grouped organisms, through phylogenetic analyses, for further study. The 23S rRNA gene sequences are more reliable, allowing for a more robust, reproducible and accurate bacterial identification than with the 16S rRNA gene.
Many other genomic regions have also been used to examine the phylogenetic relationships among bacteria. Whole-genome analyses have been tried, but these are still quite difficult because the genomes are of different sizes and gene duplication, transfer, deletion, fusion and splitting are common in them9,21,22. However, it has been observed that the trees based on genomic data and 23S rRNA gene data are similar, showing that 23S rRNA sequences can aid in distinguishing between Streptomyces spp.
Several observations can be made from comparing the concatenated eight-gene and the 16S rRNA gene trees. First, the phylogenetic relationships between most strains in this study are usually different between the two trees. Second, the eight-gene tree shows a much higher power of discrimination since most species are clearly discriminated from each other in it. Third, the topological structure of the eight-gene tree, which is supported by noticeably higher bootstrap values, is much more stable than that of the 16S rRNA gene tree, a similar behavior to that observed by Rong and Huang7. These observations emphasize the fact that the eight-gene tree is obviously superior to the 16S rRNA gene tree in both resolution power and topological stability. Phylogenetic trees predicted from each of the forty genes were varied, however, with slight differences, thus forming the basis for the concatenation of the eight chosen genes for the analysis (Fig. 2). The phylogenetic tree based on the eight concatenated genes had a similar topology to the 23S rRNA gene tree (Fig. 3), with most of the branches obtained with the 23S rRNA gene recovered, but with better discriminated entities and higher bootstrap support values. The results using the internal fragment of 23S for the 26 strains demonstrated that the phylogeny was generally congruent with that of the whole gene region and showed much higher power of discrimination and a stable topological structure. Although a few branches presented dissimilar taxa, it is still more cost-effective using this partial gene sequence of 23S for everyday use in identification of novel isolates. This technique is of great biological significance since it provides a tool that will benefit both ecology and bioprospecting of these ubiquitous microorganisms. With a primer pair designed for a partial 23S (F1067 5’-GGGGATAAGCTCCATGGTCG3’ and R2192 5’-AAGTTCTCAGCTTCGCCAC-3’ (Tm 58)), it was possible to amplify all Streptomyces strains in our laboratory.
Understanding the extent of genetic and functional diversity among strains of the same or very closely related species has become a cornerstone issue for bacterial systematic23,24. With the availability of several genomes, it has become more attractive to survey the diversity and evolution of bacteria, since it assists in phylogenetic reconstruction and evolutionary studies by providing larger numbers of informative characters, which in turn allows for comparisons between the history and changes in the genes present in the genome15,25.
The 23S rRNA gene phylogeny of Streptomyces can predict the diversity of secondary metabolites from strains, further supporting the diversity identified by genome fingerprinting. Moreover, this result demonstrated another advantage of 23S, since it behaves like the secondary metabolic pathway genes, it can give an appropriate prediction of relatedness and diversification of organisms. With the decrease of sequencing costs, partial sequences of the 23S rRNA gene can be a source of data for both systematic research and functional investigation. With the availability of whole genome sequences, they can be used for defining the true relationships between Streptomyces species. The concatenation approach has been used by a number of phylogenetic studies6,7,15. The phylogenies obtained from the 23S gene are of great biological significance since they provide an elaborate taxonomic grouping of streptomycetes and will benefit both ecology and bioprospecting of these ubiquitous microorganisms.
CONCLUSION
In this study developed a 23S rRNA gene sequence analysis scheme for Streptomyces and have shown its promising potential for refining the phylogeny of this genus. The scheme was based on an internal fragment of the 23S rRNA gene and can discriminate and define phylogenetic relationships between diverse and closely related species of Streptomyces. This can be a valuable tool in the discovery of novel and commercially important metabolites.
SIGNIFICANCE STATEMENTS
Streptomyces is one of the most important sources of bio-active molecules for medicine and industry. There is currently a difficulty in identifying Streptomyces at the species level, especially when the 16S rDNA gene is used, there is a need to discover other genes that may better represent the diversity of this important genus. This study will help researchers identify Streptomyces strains more accurately and better organizing this important genus of bacteria, using the 23S rDNA gene.
ACKNOWLEDGMENTS
The authors would like to thank Universidade do Estado do Amazonas, UEA for financial support. This study was supported by grants MBT/UEA2015 from CAPES.
REFERENCES
- Procopio, R.E.D.L., I.R. da Silva, M.K. Martins, J.L. de Azevedo and J.M. de Araujo, 2012. Antibiotics produced by Streptomyces. Brazil. J. Infect. Dis., 16: 466-471.
CrossRefDirect Link - Bentley, S.D., K.F. Chater, A.M. Cerdeno-Tarraga, G.L. Challis and N.R. Thomson et al., 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature, 417: 141-147.
CrossRefDirect Link - Wang, X.J., B. Zhang, Y.J. Yan, J. An and J. Zhang et al., 2013. Characterization and analysis of an industrial strain of Streptomyces bingchenggensis by genome sequencing and gene microarray. Genome, 56: 677-689.
CrossRefPubMedDirect Link - Alam, M.T., M.E. Merlo, E. Takano and R. Breitling, 2010. Genome-based phylogenetic analysis of Streptomyces and its relatives. Mol. Phylogenet. Evol., 54: 763-772.
CrossRefPubMedDirect Link - Harrison, J. and D.J. Studholme, 2014. Recently published Streptomyces genome sequences. Microb. Biotechnol., 7: 373-380.
CrossRefDirect Link - Guo, Y., W. Zheng, X. Rong and Y. Huang, 2008. A multilocus phylogeny of the Streptomyces griseus 16S rRNA gene clade: Use of multilocus sequence analysis for streptomycete systematics. Int. J. Syst. Evol. Microbiol., 58: 149-159.
CrossRefDirect Link - Rong, X. and Y. Huang, 2010. Taxonomic evaluation of the Streptomyces griseus clade using multilocus sequence analysis and DNA-DNA hybridization, with proposal to combine 29 species and three subspecies as 11 genomic species. Int. J. Syst. Evol. Microbiol., 60: 696-703.
CrossRefPubMedDirect Link - Chater, K.F. and G. Chandra, 2006. The evolution of development in Streptomyces analysed by genome comparisons. FEMS Microbiol. Rev., 60: 651-672.
CrossRefPubMedDirect Link - Colston, S.M., M. Fullmer, L. Beka, B. Lamy and J.P. Gogarten and J. Graf, 2014. Bioinformatic genome comparisons for taxonomic and phylogenetic assignments using Aeromonas as a test case. mBio, Vol. 5, No. 6.
CrossRefDirect Link - Land, M., L. Hauser, S.R. Jun, I. Nookaew and M.R. Leuze et al., 2015. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genomics, 15: 141-161.
CrossRefDirect Link - Stothard, P., 2000. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques, 28: 1102-1104.
PubMedDirect Link - Tamura, K., D. Peterson, N. Peterson, G. Stecher, M. Nei and S. Kumar, 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol., 28: 2731-2739.
CrossRefPubMedDirect Link - Felsenstein, J., 1985. Phylogenies and the comparative method. Am. Natl., 125: 1-15.
CrossRefDirect Link - McWilliam, H., W. Li, M. Uludag, S. Squizzato and Y.M. Park et al., 2013. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res., 41: W597-W600.
CrossRefPubMedDirect Link - Rong, X., N. Liu, J. Ruan and Y. Huang, 2010. Multilocus sequence analysis of Streptomyces griseus isolates delineating intraspecific diversity in terms of both taxonomy and biosynthetic potential. Antonie Van Leeuwenhoek, 98: 237-248.
CrossRefPubMedDirect Link - Mori, H., F. Maruyama and K. Kurokawa, 2010. VITCOMIC: Visualization tool for taxonomic compositions of microbial communities based on 16S rRNA gene sequences. BMC Bioinf., Vol. 11, No. 332.
CrossRefDirect Link - Mizrahi-Man, O., E.R. Davenport and Y. Gilad, 2013. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: Evaluation of effective study designs. PLoS ONE, Vol. 8, No. 1.
CrossRefDirect Link - Kim, B.J., C.J. Kim, J. Chun, Y.H. Koh and S.H. Lee et al., 2004. Phylogenetic analysis of the genera Streptomyces and Kitasatospora based on partial RNA polymerase β-subunit gene (rpoB) sequences. Int. J. Syst. Evol. Microbiol., 54: 593-598.
CrossRefDirect Link - Clarridge, J.E., 2004. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev., 17: 840-862.
CrossRefDirect Link - Alfoldi, J. and K. Lindblad-Toh, 2013. Comparative genomics as a tool to understand evolution and disease. Genome Res., 23: 1063-1068.
CrossRefDirect Link - Bansal, A.K. and T.E. Meyer, 2002. Evolutionary analysis by whole-genome comparisons. J. Bacteriol., 184: 2260-2272.
CrossRefDirect Link - Koeppel, A., E.B. Perry, J. Sikorski, D. Krizanc and A. Warner et al., 2008. Identifying the fundamental units of bacterial diversity: A paradigm shift to incorporate ecology into bacterial systematics. Proc. Natl. Acad. Sci. USA., 105: 2504-2509.
CrossRefDirect Link - Bohlin, J., O.B. Brynildsrud, C. Sekse and L. Snipen, 2014. An evolutionary analysis of genome expansion and pathogenicity in Escherichia coli. BMC Genomics, Vol. 15.
CrossRefDirect Link - Bentley, S.D. and J. Parkhill, 2015. Genomic perspectives on the evolution and spread of bacterial pathogens. Proc. Biol. Sci., Vol. 282.
CrossRefDirect Link