
N.A. Wahab
Faculty of Applied Sciences, Universiti Teknologi MARA, 40450 Shah Alam, Selangor, Malaysia
Z.M. Noor
Faculty of Applied Sciences, Universiti Teknologi MARA, 40450 Shah Alam, Selangor, Malaysia
M.F.F. Abdullah
Faculty of Applied Sciences, Universiti Teknologi MARA, 40450 Shah Alam, Selangor, Malaysia
Research Journal of Microbiology
Year: 2012 | Volume: 7 | Issue: 1 | Page No.: 41-49







ABSTRACT
An E. coli yeast shuttle vector for the anchoring of heterologous protein to the yeast host’s cell wall was constructed. The vector PYDSM01 includes a DNA sequence constructed from the signal sequence from the yeast sucrose isomerase gene, a multiple cloning site and a DNA fragment encoding the carboxyl-terminal of the yeast cell wall protein 2 (CWP2). This construct was then inserted into the HindIII site on pGAD424, replacing the GAL4 fusion tag and the original MCS sequence. DNA sequencing confirmed the correct insertion of both signal and anchor proteins in the vector. To test for proper expression and functional anchoring to the cell wall, the coding sequence for a bacterial alpha-amylase enzyme was cloned into the vector and transformed into a yeast host. A total of 22 yeast transformant were recovered which were able to degrade starch, indicating successful expression and function of the bacterial alpha-amylase gene. Enzyme assay of the washed cell pellet and supernatant fractions indicate that the both activity and anchoring efficiency are variable.
PDF Abstract XML References Citation
Received: August 04, 2011;
Accepted: November 23, 2011;
Published: January 14, 2012
How to cite this article
N.A. Wahab, Z.M. Noor and M.F.F. Abdullah, 2012. Construction of a Vector for the Surface Display of Heterologous Proteins in Saccharomyces cerevisiae. Research Journal of Microbiology, 7: 41-49.
URL: https://scialert.net/abstract/?doi=jm.2012.41.49
URL: https://scialert.net/abstract/?doi=jm.2012.41.49
INTRODUCTION
The use of the yeast Saccharomyces cerevisiae to display heterologous-expressed proteins on the cell surface was pioneered two decades ago (Schreuder et al., 1993). In the early phases, yeast surface display was used mainly for immobilization of enzymes (Pepper et al., 2008). A variety of enzymes such as amylase and glucoamylase (Schreuder et al.,1993; Murai et al., 1997a), cellulolytic enzymes (Murai et al., 1997b; Murai et al., 1998; Fujita et al., 2002) and lipase (Matsumoto et al., 2004) has been successfully displayed on the yeast cell wall. More recently, several groups have successfully demonstrated the simultaneous anchoring of multiple enymes e.g., β-glucosidase and carboxymethylcellulase (Murai et al., 1998) and also glucoamylase and α-amylase of S. diastaticus and B. amyloliquefaciens origin respectively in the same yeast cell (Steyn and Pretorius, 1991). Co-expression of glucoamylase of Rhizopus oryzae and α-amylase of B. stearothermophilus (Murai et al., 1999) were further found to enhance the stability of both enzymes. Surface display permits more than single modification to be carried-out on the subject protein and some has reported a synergistic combination of protein engineering and display system e.g., when lipase of R. oryzae origin was displayed on cell surface of S. scerevisiae in organic solvents, the catalytic activity was several folds higher than the original construct (Shiraga et al., 2005). Functional minicellulosome complexes consisting of endoglucanase, cellobiohydrolase and β-glucosidase arranged on a scaffold molecule has been expressed and assembled on the surface of yeast, enabling the simultaneous breakdown and fermentation of cellulose to ethanol (Pepper et al., 2008).
Besides enzymes, pioneering work by Boder and Wittrup (1997) used yeast surface display to create combinatorial protein libraries of human antibody Fab fragments for affinity selection. This was further developed to include application for other human proteins with therapeutic potential e.g., T-cell receptors (Pepper et al., 2008). Other applications include the displaying of fluorescent protein as a reporter gene (Shibasaki et al., 2001), protein A (SpA) for immunoglobulin binding (Nakamura et al., 2001), a histidine oligopeptide (Hexa-His) for chelating heavy metal ions (Kuroda and Ueda, 2003; Kuroda et al., 2001) and the endocrine disruptor group of proteins (Routledge and Sumpter, 1997) for environmental screening of waste water.
Early methods for anchoring proteins onto the yeast’s cell wall were achieved by using either the β-agglutinin Aga2p or flocculin Flo1p as the cell wall anchor protein. The Aga2p system, developed by Boder and Wittrup (1997) anchors the fusion protein by disulfide bonding to Aga1p on the yeast’s cell wall. The Flo1p protein is used when non-covalent display is desired (Kondo and Ueda, 2004). The Pir family of cell wall proteins was also used to display human glycosyltransferase enzymes (Shimma et al., 2006). The selection of a suitable anchor appears to play an important role in successful expression and functionality of the fusion protein (Abe et al., 2003). Earlier work by Van der Vaart et al. (1997) compared the effectiveness if several yeast cell wall proteins Cwp1p, Cwp2p, Aga1p, Tip1p, Flo1p, Sed1p, YCR89W and Tir1p in functionally displaying an β-galactosidase enzyme and concluded that a the C-terminal fragment of Cwp2p was the most effective anchor. Breinig and Schmitt (2002) used Cwp2p to express the haemagglutinin (HA) epitope, and subsequently a bacterial esterase (Breinig et al., 2006). Besides these, however, Cwp2p was not widely used as an anchor.
While the technology for yeast surface display have been taken to a new level of sophistication, there is still a need for a simple system capable of displaying proteins of industrial interest e.g., enzymes at high density, while being firmly bonded to the cell. The most widely used Aga2p system relies on disulfide bonds between Aga1p and Aga2p for attachment. For this purpose, we design a vector for yeast surface expression using the 67 C-terminal amino acids of Cwp2p as an anchor.
MATERIALS AND METHODS
Construction of pYDSM01: The backbone of surface-display vector was derived from plasmid pGAD424, a yeast two-hybrid vector (Clontech, USA). The GAL4 AD domain fusion tag and Multiple Cloning Site (MCS) of pGAD424 was excised by digestion with HindIII. This resulted in a linearised plasmid backbone with HindIII overhangs. The two fragments were separated on a 0.8% agarose gel by electrophoresis and the backbone recovered by band excision and extraction using a commercial kit (Qiagen, USA). The linearised plasmid was then dephosphorylated using Antartic phosphatase (NEB).
A two-step PCR strategy was used to build the cloning construct (Fig. 1). Primers YS1F 5’- ATCGAGAATTCCCGGGGATCCGTCGACCTGCAGAGATCTATATTTCTCAAATCACTGAC GGTC and YS2R 5’- ATCGAAAGCTTTTATAACAACATAGCAGCAGCAG were used to amplify a fragment coding for 67 amino acids on the C-terminal of CWP2. The first 30 nucleotides of the forward primer YS1F contain part of the signal sequence for the yeast sucrose isomerase and a Multiple Cloning Site (MCS) sequence. Yeast genomic DNA was extracted using QIAGEN’s Dneasy kit and used as the target DNA.
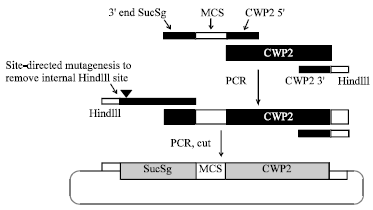 | |
| Fig. 1: | Schematic diagram for vector construction (Wahab et al., 2011). (SucSg = Signal sequence of sucrose isomerase) |
The required fragment was generated by Polymerase Chain Reaction (PCR) with a denaturation temperature of 94°C for 15 sec, annealing at 56°C for 30 sec, extension at 68°C for 1 min, for 30 cycles on an Eppendorf Mastercycler. The resulting amplification product from this amplification step contains part of the 3’ end of the sucrose isomerase signal sequence, an in-frame MCS and sequences coding for the N-terminal 67 amino acids of Cwp2p.
A second set of primers was used to make the complete insert. The forward primer (YS3F) consists of the first 40 nucleotides of the sucrose isomerase signal sequence. The internal HindIII site in the signal sequence was abolished by site-directed mutagenesis by substituting A for T in the fifth codon. The resulting mutation is silent. A flanking HindII site was added immediately before the start codon. The reverse primer was YS2R. The PCR product was purified and verified by sequencing. The fragment was then digested with HindIII and ligated into the linearised pGAD424 backbone. Post ligation, the reaction mix was transformed into E.coli DH5 cells. Screening was carried-out by plating on Luria-Bertani (LB) agar plates supplemented with ampicillin (100 mg L-1). The resulting vector (PYDSM01) was recovered from positive transformants and verified by restriction enzyme mapping and DNA sequencing.
Cloning of the bacterial α-amylase gene: Genomic DNA was extracted from Bacillus subtilis ATCC 6633 using Qiagen’s DNeasy kit and used as the source of bacterial amylase gene. PCR was carried out using primers BA1F and BA2R targeting the entire ORF minus the start and stop codons. PCR was performed at 94°C for 15 sec, annealing at 55°C for 30 sec, extension at 68°C for 3 min, for 30 cycles on an Eppendorf Mastercycler. The amplification product was purified using a commercial kit and sequenced for verification. The purified PCR product was digested with BamHI and cloned into pYDSM01 at the BamHI site in the MCS. Post ligation, the reaction mix was transformed into E.coli DH5α cells as described above. Plasmids were recovered from positive transformants and the insertion was verified by sequencing. For expression and surface display, the recombinant plasmid was transformed into a leu2 yeast strain using the Lithium acetate method of Gietz (1992). Putative transformants were selected on synthetic complete media lacking leucine and characterized.
Cloning the GFP gene: The coding sequences for a variant of the green fluorescent protein optimized for expression in yeast was amplified from plasmid pFA6a-GFP-KanMX (a gift from R.Borts, U.Leicester). The amplified sequence were purified, sequenced, digested with BamHI and inserted into pYDSM01 as described above. Transformants were selected on synthetic complete media lacking leucine and characterized.
Characterization of yeast transformants: Yeast transformants were cultured individually in 96-well plates in 100 μL of YEPD. After overnight growth at 30°C, 100 μL of 1% starch solution were added to each well and incubated for 1 h at 30°C. A drop of Gram’s iodine was added to detect for residual starch. Transformants with amylase activities were selected, inoculated into YEPD supplemented with 100 mg L-1 ampicillin to prevent bacterial contamination and grown overnight at 30°C. The cells were then recovered by centrifugation, washed twice and resuspended in phosphate buffer (pH 7). Both the washed cells and the culture supernatant were then assayed for amylase activity by testing for the production of free glucose from starch using the AMPLEX Glucose kit (Invitrogen, USA). Amylase activity was measured in three replicates for each transformant and the results averaged.
For transformants carrying the GFP insert, cells grown overnight in YEPD were harvested by centrifugation, washed twice and resuspended in phosphate buffer. An aliquot was mounted in antifade mountant (Invitrogen) and observed under a fluorescent microscope using appropriate filters. A control consisting of yeast cell transformed with the vector only was prepared and treated in exactly the same manner.
RESULTS AND DISCUSSION
Vector construction and gene cloning: Successful construction of vector PYDSM01 was verified by sequencing (Fig. 2). DNA sequencing confirms the correct orientation and reading frame of the insert.
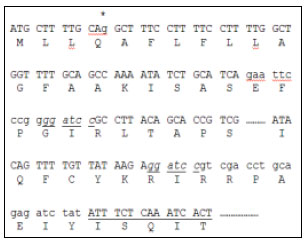 | |
| Fig. 2: | DNA sequence and corresponding amino acid sequence of the construct. The first 20 codons codes for the sucrose isomerase signal sequence. The asterisk *marks the nucleotide that was mutated to abolish HindIII restriction site. Sequences in small letters indicate part of the engineered MCS, followed part of the 5’ end of the B.subtilis alpha-amylase gene. The entire construct was sequenced and verified to be free of PCR-induced mutations. The sequences were also checked at both BamHI junctions (italics and underlined) to ascertain that the entire construct is in-frame with respect to all the three sequences. The first 6 codons of the CWP2 fragment are shown in the figure (underlined) |
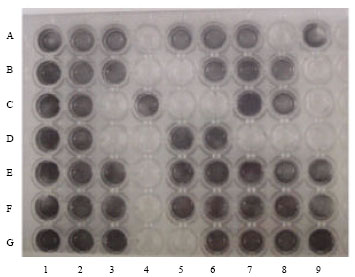 | |
| Fig. 3: | Screening for yeast transformants displaying amylase activity. Wells A1 to G1 contain yeast cells transformed with pGAD424 as controls. No amylase ac tivities were observed in these cells, as evident by the dark blue color of the starch-iodine complex. Clear wells shows reactions where the starch has been hydrolysed, indicating transformants with amylase activity |
Preliminary screening of transformants: Yeast transformants were first selected for the presence of amylase activity using a simple starch hydrolysis assay. Figure 3 shows an example of a 96-well microtiter plate screening of transformants expressing the α-amylase enzyme. The yeast host that is used does not contain an amylase gene and no amylase activity was observed in the non-transformed yeast cells (data not shown) or cells transformed with the vector only. Clear, or very light purple wells can be seen at A3,A7, B3- 4, B8, C2, C4- 5, C8, D2- 3, D6- D8, E3, F3 and G3-4. All transformants which tested positive for starch hydrolysis were selected for secondary screening. While the expression of the amylase gene appears to be a stable phenotype, the level of expression differs among the positive transformants (Fig. 4). Some transformants were able to completely hydrolyse the starch substrate within 30 min (clear wells) while in some wells a light purple color, indicative of residual starch, can still be observed at the end of the incubation period.
Glucose assay: Table 1 shows results from glucose assay of the amylase activity of the cell and supernatant fractions from selected transformants. The transformants were grown in YEPD and centrifugation was used to separate the cell and supernatant. The cell pellet was washed and resuspended in PBS.
Sample Y0 was the original yeast host transformed with the unmodified pGAD424 vector. No amylase activity was detected in either the cell or supernatant fractions. For transformants Y14 and Y22, amylase activity was detected only in the supernatant fractions. These represent transformants in which the amylase gene was successfully expressed and secreted out of the cell, but the fusion protein was not incorporated into the yeast cell wall. Possible explanation includes premature truncations of protein synthesis, leading to the loss of the Cwp2p tag. For transformants Y5, Y7 and Y9, the amylase activity was detected only on the cell pellet. These represent transformants in which the recombinant construct was successfully expressed, secreted and anchored correctly on the cell wall. Except for transfrormant Y5, the cell pellet fractions in general display lower enzyme activities compared to the supernatant.
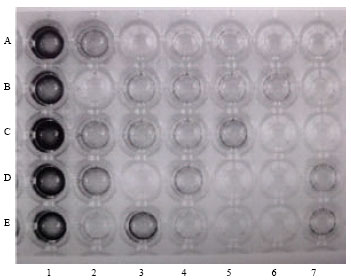 | |
| Fig. 4: | Secondary screening for yeast transformants that express amylase activities. Column A1 to E4 are cells transformed with the vector only. The expression of the alpha-amylase gene appears to be variable among positive transformants |
 | |
| Fig. 5(a-b): | Yeast cells transform with vector and GFP insert fluorescing green color when excited with blue light. In (a) the fluorescence is concentrated near the cell surface. (b) Fluorescence appears to be diffused in the cell |
It is possible that anchoring by Cwp2p somehow interferes with the amylase function.
In six transformants (Y2, C1-C5), substantial amylase activities was found on both the cell pellet and supernatant fractions. These are transformants in which the construct was successfully expressed at a high level but a portion of the amylase enzymes were not properly anchored. Similar observations were reported by Murai et al. (1997a) with fungal amylases and were hypothesized to be caused by proteolytic processing of the fusion protein, leading to loss of the anchor tag. Transformants C1-C5 was obtained from a different batch of experiment. It appears that the physiology of the host cell during transformation may have an effect on the processing of the fusion proteins.
No activity was detected from transformant Y18 which showed positive results in the preliminary screening. This was also observed in a number of transformants that lost their amylase activity during the secondary screen.
| Table 1: | Detection of amylase activity on the cell pellet and supernatant fractions of transformants. Y0 represent the average reading for more than 10 clones of yeast cells transformed with the vector only |
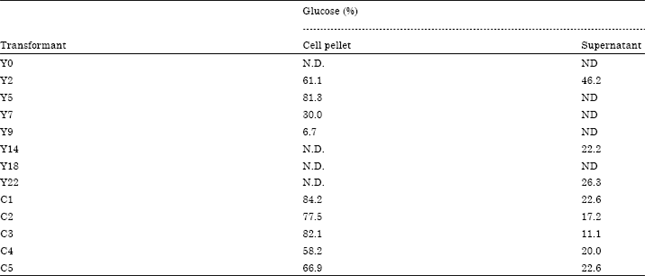 | |
| ND: Not detected | |
The recovery of three types of transformants indicate that the constructed vector pYDSM01 is able to expressed a cloned gene fused to a signal sequence and a C-terminal Cwp2p tag that anchors to the cell wall of a yeast host. However, expression appears to be variable in some transformants. Some transformants also failed to have the fusion protein displayed on the cell wall, although enzyme activity was detected, indicating successful expression but not anchoring. This variable expression and anchoring can be due to a number of factors e.g., instability of the recombinant plasmid, variable promoter activity or premature truncation of the fusion protein resulting in loss of enzyme activity or the Cwp2 tag.
It is also possible that we tried to express a bacterial enzyme, which may not be fully functional in the yeast host cell. For prokaryotic enzymes, proper folding appears to be required for maximum activity (Shimma et al., 2006).
Expression of GFP: Yeast cell transformed with the vector containing cloned GFP sequences were observed to fluoresce after overnight growth (Fig. 5). In a number of cells, the fluorescence pattern appears to be localized to the periphery of the cell, indicating that the GFP is concentrated on the cell wall. In some cells however, the green fluorescence appears to be diffuse within the cell, possibly indicating free proteins in the cytoplasm. There were also non-fluorescing cell. No fluorescence was observed in the negative control which are transformants carrying an empty vector. All samples and controls were processed in the same manner after the same period of growth.
The same variable phenotypes were thus observed for GFP as in yeast transformants expressing amylase. Thus while Cwp2p was reported to be the most efficient tag among many tested by Van der Vaart et al. (1997), the anchoring efficiency is not consistent. A certain portion of the protein appears to be able to detached from the cell surface, or failed to be transported to the cell wall and remain in the cytoplasm. This could be due to inefficiency of the sucrose isomerase signal sequence in targetting the fusion protein to the cell wall, or failure of the Cwp2p tag to anchor firmly on the cell surface. The latter could be a result of using only 67 amino acid at the C-terminal of Cwp2p, or due to interference from the fusion amylase enzyme. By adding a spacer sequence between the anchor and the tag, (Breinig and Schmitt, 2002) were able to enhance anchoring.
CONCLUSIONS
The carboxy terminal of Cwp2p was functional as an anchor protein for the expression of a bacterial enzyme. However, both expression and anchoring efficiency was variable. Further, optimization will be required before the system can be used in industrial processes.
ACKNOWLEDGMENT
The authors would like to thank the RMI, UiTM for supported work under a short term grant and the NSF, MOSTI for a scholarship to Nadzarah A.Wahab.
REFERENCES
- Abe, H., Y. Shimma and Y. Jigami, 2003. In vitro oligosaccharide synthesis using intact yeast cells that display glycosyltransferases at the cell surface through cell wall-anchored protein Pir. Glycobiology, 13: 87-95.
PubMed - Steyn, A.J. and I.S. Pretorius, 1991. Co-expression of a Saccharomyces diastaticus glucoamylase-encoding gene and a Bacillus amylo-liquefaciens amylase-encoding gene in Saccharomyces cerevisiae. Gene, 100: 85-93.
Direct Link - Boder, E.T. and K.D. Wittrup, 1997. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol., 15: 553-557.
CrossRef - Fujita, Y., S. Takahashi, M. Ueda, A. Tanaka and H. Okada et al., 2002. Direct and efficient production of ethanol from cellulosic material with a yeast strain displaying cellulolytic enzymes. Applied Environ. Microbiol., 68: 5136-5141.
PubMed - Kuroda, K. and M. Ueda, 2003. Bioadsorption of cadmium ion by cell surface-engineered yeasts displaying metallothionein and hexa-His. Applied Microbiol. Biotechnol., 63: 182-186.
PubMed - Matsumoto, T., M. Ito, H. Fukuda and A. Kondo, 2004. Enantioselective transesterification using lipase-displaying yeast whole-cell biocatalyst. Applied Microbiol. Biotechnol., 64: 481-485.
CrossRef - Murai, T., M. Ueda, M. Yamamura, H. Atomi and Y. Shibasaki et al., 1997. Construction of a starch-utilizing yeast by cell surface engineering. Applied Environ. Microbiol., 63: 1362-1366.
PubMed - Murai, T., M. Ueda, H. Atomi, Y. Shibasaki and N. Kamasawa et al., 1997. Genetic immobilization of cellulase on the cell surface of Saccharomyces cerevisiae. Applied Microbiol. Biotechnol., 48: 499-503.
PubMed - Murai, T., M. Ueda, T. Kawaguchi, M. Arai and M. Tanaka, 1998. Assimilation of cellooligosaccharides by a cell surfaceengineered yeast expressing β-glucosidase and carboxymethylcellulase from Aspergillus aculeatus. Appl. Environ. Microbiol., 64: 4857-4861.
Direct Link - Murai, T., M. Ueda, Y. Shibasaki, N. Kamasawa, M. Osumi, T. Imanaka and A. Tanaka, 1999. Development of an arming yeast strain for efficient utilization of starch by co-display of sequential amylolytic enzymes on the cell surface. Applied Microbiol. Biotechnol., 51: 65-70.
PubMed - Nakamura, Y., S. Shibasaki, M. Ueda, A. Tanaka, H. Fukuda and A. Kondo, 2001. Development of novel whole-cell immunoadsorbents by yeast surface display of the IgG-binding domain. Applied Microbiol. Biotechnol., 57: 500-505.
PubMed - Pepper, L.R., Y.K. Cho, E.T. Boder and E.V. Shusta, 2008. A decade of yeast surface display technology: Where are we now? Comb. Chem. High Throughput. Screen., 11: 127-134.
PubMed - Routledge, E.J. and J.P. Sumpter, 1997. Structural features of alkylphenolic chemicals associated with estrogenic activity. J. Biol. Chem., 272: 3280-3288.
Direct Link - Schreuder, M.P., S. Brekelmans, H. Van Den Ende and F.M. Klis, 1993. Targeting of a heterologous protein to the cell wall of Saccharomyces cerevisiae. Yeast, 9: 399-409.
CrossRef - Shibasaki, S., M. Ueda, K. Ye, K. Shimizu, N. Kamasawa, M. Osumi and A. Tanaka, 2001. Creation of cell surface-engineered yeast that display different fluorescent proteins in response to the glucose concentration. Applied Microbiol. Biotechnol., 57: 528-533.
CrossRef - Shimma, Y., F. Saito, F. Oosawa and Y. Jigami, 2006. Construction of a library of human glycosyltransferases immobilized in the cell wall of Saccharomyces cerevisiae. Applied Environ. Microbiol., 72: 7003-7012.
CrossRef - Shiraga, S., M. Kawakami, M. Ishiguro and M. Ueda, 2005. Enhanced reactivity of Rhizopus oryzae lipase displayed on yeast cell surfaces in organic solvents: Potential as a whole-cell biocatalyst in organic solvents. Applied Environ. Microbiol., 71: 4335-4338.
CrossRef - Van der Vaart, J.M., R. te Biesebeke, J.W. Chapman, H.Y. Toschka, F.M. Klis and C.T. Verrips, 1997. Comparison of cell wall proteins of Saccharomyces cerevisiae as anchors for cell surface expression of heterologous proteins. Applied Environ. Microbiol., 63: 615-620.
Direct Link - Gietz, D., A. St Jean, R.A. Woods and R.H. Schiestl, 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res., 20: 1425-1425.
CrossRefPubMedDirect Link