
Humodi A. Saeed
College of Medical Laboratory Science, Sudan University of Science and Technology, Sudan
Zahra K. Yousif
National Health Laboratory, Khartoum, Sudan
Mugahid M. El Hassan
College of Medical Laboratory Science, Sudan University of Science and Technology, Sudan
Misk El Yamen A. Atti
College of Medical Laboratory Science, Sudan University of Science and Technology, Sudan
Mansour M. Mansour
College of Medical Laboratory Science, Sudan University of Science and Technology, Sudan
Research Journal of Microbiology
Year: 2009 | Volume: 4 | Issue: 4 | Page No.: 173-177







ABSTRACT
This study was undertaken in Khartoum State, Sudan, during the period May 2007 to March 2008. A detection system based on real-time PCR has been developed for detection of Escherichia coli strains in patients’ urine. The optimized assay format included two PCR primers. Urine specimens (46) were collected from patients attending different hospitals in Khartoum State. Bacterial DNA was extracted from each urine specimen using the Phenol-Chloroform method. Real time PCR technique was adopted to detect E. coli. The study revealed that 45.7% of the specimens were positive for E. coli. The bacterium was more prevalent in female patients than in male patients. Adult age group was more exposed to the pathogen than the children age group. Real-time PCR technique facilitated detection of E. coli directly in patients’ urine without a need for bacterial culture. The technology could be easily adopted in hospital settings in the Sudan.
PDF Abstract XML References Citation
How to cite this article
Humodi A. Saeed, Zahra K. Yousif, Mugahid M. El Hassan, Misk El Yamen A. Atti and Mansour M. Mansour, 2009. A Real-Time Polymerase Chain Reaction Based Assay for the Detection of Escherichia coli in Patients with Urinary Tract Infection in the Sudan. Research Journal of Microbiology, 4: 173-177.
URL: https://scialert.net/abstract/?doi=jm.2009.173.177
URL: https://scialert.net/abstract/?doi=jm.2009.173.177
INTRODUCTION
Escherichia coli (E. coli)is the prototype of the large bacterial family Enterobacteriaceae. It is facultatively anaerobic with both fermentative and respiratory type of metabolism. E. coli is one of the most frequent causes of some of the many common bacterial infections of man such as urinary tract infection, neonatal meningitis, choleycystitis, bacteremia, cholangitis, traveler’s diarrhea and pneumonia (Ochoa and Cleary, 2003). In the past the isolation of E. coli was done by simple methods. Clinical specimens may be stained by Gram’s method for microscopical examination or cultured on MacConkey’s agar or other suitable media. In the case of suspected urinary tract infection, cultures are semi quantitative (Chart, 1998).
Much of the past practice and thinking about E. coli were based on the classical views and behavior of the organism. These views are changing rapidly under the influence of accumulating fundamental molecular knowledge. Currently, molecular techniques are finding an increasing use in the diagnoses of E. coli. The most widely used method is the Polymerase Chain Reaction (PCR). Not only does this technique provide tools for highly sensitive and specific detection of the organism in clinical specimens, but certain characteristics including virulence, toxins and antimicrobial resistant genes may also be determined (Ram et al., 2008). Compared with the classical urine culture methods, PCR is more rapid and can detect smaller number or fragments of bacteria; which would otherwise undetectable (Yoshimasa, 2002).
A number of modifications have been made to the standard PCR reaction the most important of which is the real-time PCR which has expanded the use of the technique and broadened the spectrum of the microorganism that may be detected. The technique, in addition to being a closed system, is highly sensitive, rapid and accurate (Campbell, 2003).
The present study was designed, essentially, to establish real-time PCR for the detection of E. coli directly in clinical specimens in the Sudan.
MATERIALS AND METHODS
This study was conducted during the period May 2007 to March 2008. A total of 46 patients (24 males and 22 females) attending Khartoum, Omdurman and Khartoum North Teaching Hospitals and the National Health Laboratory suffering from urinary tract infection were enrolled in this study. Urine specimens (n = 46) were collected from each patient. The age of patients ranged between five years to eighty years. Urine specimens were collected in sterile containers, without preservatives, transported to the laboratory and immediately processed.
DNA Extraction
Bacterial DNA was extracted directly from each urine specimen using the Phenol-Chloroform method as described by Snounou et al. (1993) with some modifications. Five milliliter of urine and 10 mL of red cell lysis buffer (RCLB) were transferred under aseptic conditions and centrifuged at 6000 rpm for 5 min. The supernatant was discarded and the deposit was re-suspended in 800 μL of white blood cell lysis buffer (WCLB) containing 10 μL of Proteinase k (10 mg mL-1) and incubated overnight at 37°C. Equal volume of phenol/chloroform/isoamyl alcohol (PCI) was added. The suspension, mixed thoroughly on a vortex shaker, was centrifuged at 6000 rpm for 5 min. The upper layer was transferred to a clean eppendorf tube and an equal volume of chloroform/isoamyl alcohol (CI) was added, mixed on a vortex shaker and centrifuged for 5 min at 6000 rpm. The upper layer was transferred to a clean eppendorf tube, 2 volumes of 95% cold ethanol and 1:10 of sample volume of 3M Na acetate were added and the mixture was incubated at -20°C over night prior to centrifugation for 10 min at 12000 rpm. The supernatant was discarded and the pellet was re-suspended in 8.2 mL of 70% ethanol. The suspension was centrifuged for 7 min at 12000 rpm and the supernatant was discarded. The last step was repeated, the supernatant was discarded and the pellet was air dried for 15 min, dissolved in sterilized distilled water (100 μL) and stored at -20°C till used.
Real-Time PCR
The DNA amplification and analysis were carried out using Thermocycler (Techne-Quantica). One set of primer (Left Primer 5’AGGCAGCAAATGAATTACGC ’3 and Right Primer 5’AGCCTGTTGACGCTCTTCAT 3’) was used. 2X sensimix NORef DNA Kit comprising 2Xsensimix NORef that contains reaction buffer, heat- activated Taq DNA polymerase, dNTPs, 6 Mm MgCl2, stabilizers and SYBER Green dye was utilized in this study. For a 100 reaction/plate, sterilized distilled water, 2X sensimix, 100 μL forward primer, reverse primer and SYBER Green dye were mixed in sterile eppndorf tube under sterile condition (Clean Bench–D Lab Tech). The reagents were added according to manufacturer’s recommendation with some modifications as follows: 500 μL H2O, 1250 μL 2X sensimix, 3-100 μL forward primer, 100 μL reverse primer and 5-50 μL SYBER Green dye. The plate was prepared as follows: 20 μL of master mix was placed on the wall of each well using automatic pipette, and a 5 μL aliquot of sample was placed on the other wall of the same well. Samples were made in duplicate. E. coli genomic DNA and distilled water (5 μL each) were added in two wells as a positive and negative control respectively for comparison. Finally, the plate was sealed by a sealing machine (Thermosealer-AB Gene-Combi Ltd.). For DNA amplification, the Thermocycler was programmed to denaturation at 95°C for 600 sec, amplification at 95°C for 30 sec, annealing at 58°C for 30 sec and extension step at 72°C for 30 sec).
RESULTS AND DISCUSSION
Forty six specimens were collected from patients attending Khartoum Teaching Hospital, Omdurman Teaching Hospital, Khartoum North Teaching Hospital and National Health Laboratory (NHL). The majority of specimens 20 (43.5%) were obtained from NHL and so the positive specimens 13(61.8%) (Table 1).
| Table 1: | Distribution of positive results according to the hospital |
 | |
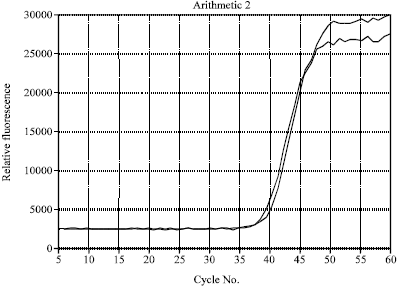 | |
| Fig. 1: | Real-time PCR curve for positive control |
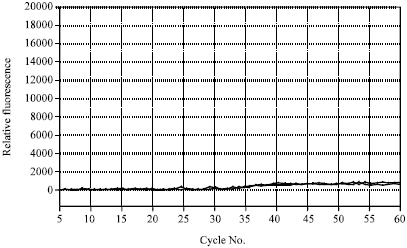 | |
| Fig. 2: | Real-time PCR curve for negative control |
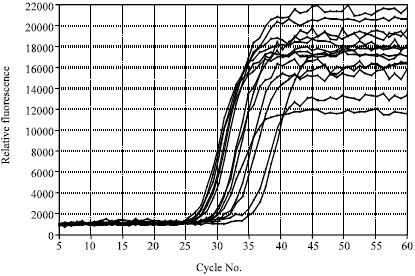 | |
| Fig. 3: | Real-time PCR sigmoid curves for specimens showing positive infection by E. coli |
| Table 2: | Distribution of positive results according to sex |
 | |
| Table 3: | Distribution of positive results according to the age groups |
 | |
Using real time PCR the overall results showed that 45.7% of the cases examined were positive for E. coli with relative fluorescence >12,000 (Fig. 3) and the rests were negative with no relative fluorescence detected (Fig. 1, 2). The number of positive samples for females and males were 13 (61.9%) and 8 (38.1%), respectively (Table 2).
The study clearly indicate the prevalence of E. coli among urinary tract infected patients. This finding is in line with that of Alizadeh et al. (2007) who reported that 39% of the urinary tract infections were caused by E. coli. Similar results were reported by Hinata et al. (2004), who ascertained the high prevalence (84%) of E. coli among urinary tract infected patients. Furthermore, our findings are in conformity with those of Hinata et al. (2004), who indicated a higher prevalence of E. coli in urine collected from female urinary tract infected patients.
Our results taken in conjunction with those of Alizadeh et al. (2007) and Hinata et al. (2004) suggested that E. coli is a major causative agent of urinary tract infection at least in many parts of the world.
The study of Hinata et al. (2004), examined 200 urinary tract infected patients, who showed a close similarity in E. coli prevalence levels using real- time PCR and the conventional culture technique. However, real- time PCR is advantageous as it is simple, more rapid, highly sensitive and more quantitative than the conventional culture techniques in the diagnosis of E. coli UTI.
Based on the results of this study it could be concluded that urine samples collected from patients attending the National Health Laboratory had the highest frequency of E. coli infection (61.8%). The age group of adult is more exposed to E. coli infection (56.5%), while the age group of children is less exposed (13.1% E. coli infection) (Table 3). The real-time PCR technique is more sensitive, specific, rapid and it can easily be adopted as a routine work in hospitals settings in the Sudan.
REFERENCES
- Alizadeh, A.H.M., N. Behrouz, S. Salmanzadeh, M. Ranjbar and M.H. Azimian et al., 2007. Escherichia coli, Shigella and Salmonella species in acute diarrhoea in Hamedan, Islamic Republic of Iran. East. Mediterr. Health J., 13: 243-249.
Direct Link - Hinata, N., T. Shirakawa, H. Okada, K. Shigemura, S. Kamidono and A. Gotoh, 2004. Quantitative detection of Escherichia coli from urine of patients with bacteriuria by real-time PCR. Mol. Diagnosis, 8: 179-184.
Direct Link - Ram, S., P. Vajpayee, U. Tripathi, R.L. Singh, P.K. Seth and R. Shanker, 2008. Determination of antimicrobial resistance and virulence gene signatures in surface water isolates of Escherichia coli. J. Applied Microbiol., 105: 1899-1908.
Direct Link - Yamamoto, Y., 2002. PCR in diagnosis of infection: Detection of bacteria in cerebrospinal fluids. Clin. Diagnostic Lab. Immunol., 9: 508-514.
Direct Link - Ochoa, T.J. and T.G. Cleary, 2003. Epidemiology and spectrum of disease of Escherichia coli O157. Curr. Opin. Infect. Dis., 16: 259-263.
PubMed - Snounou, G., S. Viriyakosol, X.P. Zhu, W. Jarra and L. Pinheiro et al., 1993. High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol. Biochem. Parasitol., 61: 315-320.
PubMed
Zarizal Suhaili Reply
This article/ journal can be more specific if the author proceed the detection scheme by differentiating each spp. as well as genus with melting curve analysis instead of using amplification plots alone.
thank you.