
N. Al Haj
Department of Microbiology and Parasitology, Faculty of Medicine and Heath Sciences,
N. S. Mariana
Department of Microbiology and Parasitology, Faculty of Medicine and Heath Sciences,
A. R. Raha
Department of Bioprocess Technology, Faculty of Biotechnology and Biomolecular Sciences, University Putra Malaysia, 43400 Serdang, Selangor, Malaysia
Z. Ishak
Biotechnology Center, Mardi, P.O. Box 12301, Malaysia
Research Journal of Microbiology
Year: 2007 | Volume: 2 | Issue: 12 | Page No.: 926-932







ABSTRACT
Diarrhea is one of the leading causes of illnesses and death among children in developing countries, where an estimated 1.3 billion episodes and 4-10 million deaths occur each year in children below 5 years of age. Escherichia coli strains are among the major bacterial causes of diarrheal illness. There are now 7 classes of diarrheagenic E. coli, namely enteropathogenic E. coli (EPEC), enterohaemorrhagic E. coli (EHEC), enteroinvasive E. coli (EIEC), enterotoxigenic E. coli (ETEC), enteroaggregative E. coli (EAEC), diarrhea-associated hemolytic E. coli (DHEC) and cytolethal distending toxin (CDT)-producing E. coli. Due to the need for costly and labor-intensive diagnostic procedures, identification of diarrheagenic E. coli (DEC) is difficult at standard laboratories. Therefore, the epidemiology of DEC infections remains an important issues particularly developing country. Recently, Polymerase Chain Reaction (PCR) or dot blot has been used for genetic detection of DEC. In this study, we analyzed 25 E. coli isolates from different sources in Malaysia. Using primers for 671 bp gad gene successfully amplified by PCR. Dot blot analysis for high-throughput, rapid, simple and inexpensive quantification of specific microbial populations was evaluated and used to confirm the results of PCR. The protocol of the assays is readily applicable for implementation in the food processing, water quality control and clinical diagnosis.
PDF Abstract XML References
How to cite this article
N. Al Haj, N. S. Mariana, A. R. Raha and Z. Ishak, 2007. Development of Polymerase Chain Reaction and Dot Blot Hybridization to Detect Escherichia coli Isolates from Various Sources. Research Journal of Microbiology, 2: 926-932.
URL: https://scialert.net/abstract/?doi=jm.2007.926.932
URL: https://scialert.net/abstract/?doi=jm.2007.926.932
INTRODUCTION
Conventional microbiological methods for detection of pathogenic E. coli, however, usually include multiple subcultures and biotype or serotype identification steps and thus are laborious and time-consuming (Blackburn, 1993; Naravaneni and Jamil, 2005; Swaminathan and Feng, 1994). One of the inherent difficulties in the detection of water-food borne pathogens is that they are generally present in very low numbers (<100 cfu g-1) in the midst of up to a million or more other bacteria. These microbes may be lost among a background of indigenous microflora and substances in the foods themselves may hinder recovery or injured cells and or hide in sediment. There is also the difficulty of demonstrating that the strains recovered from any sources particularly clinical and water-food sample are, indeed, pathogenic to human beings (Sockett, 1991). Existing methods for detection of pathogenic E. coli first require time-consuming biochemical procedures to determine if an isolate is E. coli, followed by molecular and cell assays to determine whether specific virulence markers are present (Grant et al., 2001). The task would be enormous if one contemplates the monitoring of 100 of pathogens on a routine basis in water and environmental samples. E. coli is an indicator of recent fecal contamination recommended by the US Environmental Protection Agency universally to be used for monitoring the microbiological quality of water, a sensitive measure of fecal pollution since it is common to almost all warm-blooded animals, including humans (USEPA, 1986, 2005). Therefore, indicators of fecal pollution were much needed (Naravaneni and Jamil, 2005).The use of E. coli as an indicator of fecal contamination relies on the assumption that its presence in water is a direct evidence of fecal contamination and indicates the possible presence of pathogens (McLellan, 2004). There is currently no procedure for isolation of E. coli from food or water based on a characteristic unique to the species and found in all virulence subgroups. In order to control outbreaks caused by food or water-borne E. coli it is important that this process be conducted rapidly and identification of a diagnostic marker to the species which can be get in all groups would be valuable. The enzyme glutamate decarboxylase (GAD) reported to be limited to E. coli and is encoded by two virtually identical genes, gadA and gadB (McDaniels et al., 1996; Grant et al., 2001; Smith et al., 1992). Objective of this study is to develop PCR and probe to detect the E. coli from different sources simultaneously.
MATERIALS AND METHODS
Sources of Sampling
Twenty five E. coli isolates as clinical, marine, river, food and animal were studied from 5 different sources in Malaysia 2003-2004. The clinical samples were stock culture of Microbiology Laboratory there were collected from Kula Lumpur Hospital (HKL), where marine and river isolates were collected from Costrica beach, Sunggi linggi river Nergeri Semblan State. The food sample was selected randomly from different restaurant in Seri Serdange area, Selangor state. The last samples of animal source were provided by bacteriology department, Faculty of Veterinary University Putra Malaysia (UPM). Water samples were collected in 500 mL sterile containers, refrigerated at 4°C and proceeds applicant within 8 h of collection in adherence to methods for E. coli enumeration (USEPA, 1986). All samples were grown on selective and differential Chromocult Coliform Agar (Merek, Germany). Confirmation was achieved by monitoring acidification and gas production during growth in Kligler Iron Agar (KIA) and Sulfide Indole Motility (SIM) media (Oxoid. UK). The presence of E. coli was confirmed by demonstration of indole production from SIM.
DNA Preparation
E. coli isolates grown on Chromoocult Coliform Agar (Merek. Germany) overnight at 37°C. A single colony of each strain was transferred to Luria-Bertani medium (Oxoid UK) and grown overnight in a 37°C shaking water bath. DNA was prepared with a DNA isolation kit (Qiagen, Germany) DNA extraction according to the manufacturer’s instructions. The genomic DNA was checked for the concentrations and purities using spectrophotometer (Shimadzu 1601 Japan).
Polymerase Chain Reaction
Oligonucleotide primers used for these amplifications were identified from GenBank listed under accession numbers M84024 (gadA). Searches for optimal primer and probe were performed with the Primer Premier3 software program following the general primer designing parameters and commercially synthesized (Biosynthec In. Malaysia). The forward gad primer, corresponding to base positions 306-323, was F-GACC TGCG TTGCGTAAAT-R. The reverse gad primer, corresponding to base positions 970-976 was F-GGGCGGGAGAAGTTGAT-R. The probe sequence F-TGCTGAACTG-TTGCTGGAAG-R. PCR amplifications were performed with a (T-personal thermal cycler Germany) and GeneAmp PCR reagent kits (Biosynthec Inc. Malaysia) according to the manufacturer’s directions for the hot start technique. The amplification of gad gene was performed in a final volume of 50 μL containing 1X BST buffer (Biosynthec Inc. Malaysia), 1.8 mM MgCl2 (Biosynthec Inc. Malaysia), 200 μm dNTPs (Fermentas Life Sciences), 5 IU taq polymerase (Biosynthec Inc. Malaysia), 10 pmoles of each primer and 200 ng μL-1 DNA template. The PCR programmer steps performed were initial denaturation at 94°C for 2 min, followed by 30 cycles of amplification steps consisting of denaturation at 94°C for 1 min, annealing at 64°C for 1 min and elongation at 72°C for 2 min. The amplification was ended with final extension at 72°C for 7 min. After amplification, an aliquot of 10 μL reaction mixture was loaded into the wells of 1.4 % agarose gel and electrophoresed, then stained with ethidium bromide and image was captured under UV illumination (Alpha ImagerTM 2200, Alpha Innotech Corporation).
Dot Blots Hybridization
The dot blot assay was also used as an alternative method to verify the specificity of the above PCR assay. The chromosomal DNA was labeled with Hors Reddish Peroxidase (HRP) using the Direct Nucleic Acid Labeling Kit ECL (Amersham Pharmacia Biotech, UK) and then used as probe for hybridization to genomic DNA of E. coli isolates. For each isolates, 100 ng of chromosomal DNA was denatured by heating at 96°C for 10 min and spotted onto Hybond-N nylon membranes (Amersham Pharmacia Biotech, UK). DNA was then fixed on to the filter by UV cross-linking by incubating in a UV cross-linking chamber (UV-Cross linker UVC-500, USA) for 3 min at the room temperature. The prehybridization and hybridization temperature were both 42°C. All filters were pre-hybridized for 1 h in 5xSSC (1.5 M sodium chloride, 0.15 M sodium citrate). Hybridization was carried out overnight with heat-denatured probe. Detection was performed using the phototope-star detection kit according to the manufacturer’s instructions (Amersham Pharmacia Biotech, UK).
RESULTS
Polymerase Chain Reaction
Detection of gad gene by PCR showed that all the isolates of E. coli used in this study were gad gene positive. A single band of approximately 671 bp was observed in all the isolates tested. The single banding pattern observed (Fig. 1 and 2) for all isolates was located at position slightly above the 660 bp based on the 100 bp DNA ladder marker (MBI Fermentas).
Dot Blot Detection
Gene probe method using the dot blot technique was used to confirm the presence of E. coli specific fragment detected in PCR. The presence of E. coli specific fragment was confirmed in the dot blot technique when a positive signal for the spot was seen on the X-ray film. The hybridized signals on the x-ray film showed positive signal for all E. coli isolates (Fig. 3).
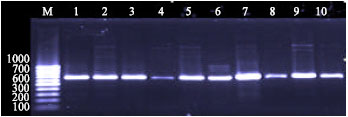 | |
| Fig. 1: | Agarose gel of electrophoresis of products amplified 671 bp gad gene, PCR from different sources of the E. coli. Lanes M: markers low molecular size standards 100 bp, Lanes 1-5: clinical and Lanes 6-10: Marine sources |
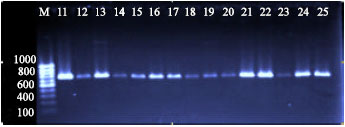 | |
| Fig. 2: | Agarose gel of electrophoresis of products amplified 671 bp gad gene, PCR from different sources of the E. coli. Lanes M: markers low molecular size standards 100 bp, Lanes 11-15: River, lane 16-20: food and Lane 21-25 animal sources |
 | |
| Fig. 3: | Photograph of the X-ray film confirming the presence of gad gene in E. coli isolates, indicated by a dot. E. coli isolates from 5 sources used in this study using design probe show positive dot blot for clinical (1-5), marine (6-10), river (11-15), food (16-20) and animal (21-25) |
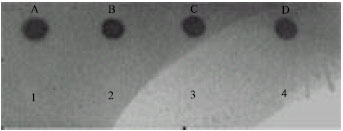 | |
| Fig. 4: | Four positive controls (A)ATCC (23519), (B) ATCC (12799), (C) ATCC (23520) and (D) ATCC (12810). 1-4 that does not show any positive signal represents (1) Vibrio cholera, (2) Salmonella sp. (3) Klebsiella pneumonia and (4) Staphylococus aureus isolates |
Specificity of E. coli Specific Probe
Specificity test by dot blot hybridization was used the Horse Radish Peroxidase (HRP) specific DNA fragment from 4 isolates of different genera of gram-positive and gram-negative bacteria (Fig. 4 and 5) Vibrio cholera, Staphylococus aureus, Salmonella sp. and Klebsiella pneumonia isolates were used as negative controls as a probe and DNA as targets was performed with E. coli isolates.
Sensitivity of E. coli Specific Probe
Probe sensitivity for E. coli was determined by hybridizing of different concentration for E. coli genomic DNA against the HRP labeled probe (Fig. 6).
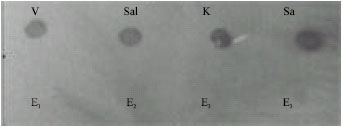 | |
| Fig. 5: | Four different genus of bacteria show positive signal for its specific probe (V) Vibrio cholera, (Sal) Salmonella sp., (K) Klebsiella pneumonia and (S) Staphylococus aureus and E1-E4 that does not show any positive signal represents E. coli |
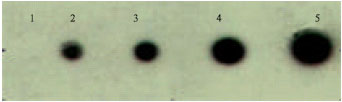 | |
| Fig. 6: | Dot 1: 50 ng of genomic DNA, dot 2-4: 100, 150, 200 and 300 ng. Dot 1 did not show any signal, dot 2 showed a faint signal, whereas from dot 3, a strong signal was observed. The detection limit of the probe developed is 100 ng. The probe developed was highly sensitive which detected as low as 100 ng of genomic DNA |
DISCUSSION
E. coli is primarily associated with food-borne diseases particularly pathogenic, contamination of drinking or recreational waters with some pathotypes has resulted in waterborne disease outbreaks and associated mortality. Identification of E. coli groups, isolates must first be identified as E. coli before they are tested for group-specific virulence traits. Because the groups are phenotypically diverse, existing methods for detecting E. coli are not always useful for pathogenic strains. Therefore we examined the potential of using gad gene as a marker for identifying E. coli isolated from various sources such as clinical and environments. PCR amplification of various sources showed that the gad gene was present in all local E. coli strains examined. There are other enteric bacteria that carried the gad genes particularly Shigella, a genus so closely related to E. coli that some investigators conclude they constitute a single species (Edwards, 1999). An ideal diagnostic test might differentiate between the two but from a public health perspective, the detection of Shigella by a GAD-based assay would not detract from the utility of this test, as Shigella is also a human pathogen because the important of E. coli as indicator of quality water which relies on the assumption that its presence in water is a direct evidence of fecal contamination and indicates the possible presence of pathogens such as Salmonella sp. Shigella sp. pathogenic E. coli and enteroviruses, including hepatitis A (McLellan, 2004). Currently, examples in the Great Lakes area include pathogenic E. coli O157:H7 isolates that contributed to a drinking water outbreak in Walkerton, Ontario, in 2000 that resulted in 2,300 illnesses and seven deaths (Hrudey and Hrudey, 2002) and to a recreational water outbreak in 2001 at a beach in Montreal, Quebec, that resulted in the hospitalization of 4 children (Bruneau et al., 2004). However, there have been few studies (Chern et al., 2004; Lauber et al., 2003; Martins et al., 1992; Obi et al., 2004) in which the proportion of potentially pathogenic E. coli isolates in the environment has been determined. Pathotyping of E. coli isolates present in water sources used for drinking or recreation could be an important tool in the development of strategies to better protect public health (Stoeckel and Harwood, 2007). Additionally, since gad-positive isolates we recommended further tested for virulence genes among E. coli isolates will be easily distinguished from pathogenic E. coli by these tests. In the present study, did not detect the presence of E. coli directly from samples without enrichment, because many foods known to contain inhibitors that can affect PCR assays (Lantz et al., 1994). Hence, culture enrichment often beneficial in diluting out the inhibitory effects of these components and is standard practice in attempts to isolate potential pathogens from contaminated food. Genomic DNA blotted on a membrane and can be detected by chemiluminescence of the hybridized signal, offers a rapid, simple, specific and accurate miniature system suitable in water-food borne area. Taking into account potentially serious water quality and food hygienic processing, rapid detection of the infectious agents based on the detection and identification the bioindicator is essential for achieving favorable management outcomes. The membrane-based assay was performed optimized to be simple and performed in 1-2 h to test E. coli genomic DNA or amplified product. The E. coli specific membrane assay developed in this study was very specific to E. coli as the probe sequence did not show positive signal for a variety of Gram positive and Gram negative bacterial tested. The sensitivity of dot blot allowed as few as 100 ng of E. coli probe hybridization assay was fast and sensitive and can be used in the laboratory methods be available for the accurate and the immediate detection of E. coli.
CONCLUSIONS
The molecular probe/primer developed and the molecular techniques optimized have led to the development of rapid molecular approach for detection E. coli isolates from various sources. We have found that the gad gene was prevalent in E. coli. DNA probe analysis showed that the gad marker was just as reliable for detecting E. coli in clinical, food and water isolates.
REFERENCES
- Blackburn, C.W., 1993. Rapid and alternative methods for the detection of salmonellas in food. J. Applied Bacteriol., 75: 199-214.
Direct Link - Bruneau, A., H. Rodrigue, J. Ismael, R. Dion and R. Allard, 2004. Outbreak of E. coli O157:H7 associated with bathing at a public beach in the Montreal-Center region. Can. Commun. Dis. Rep., 30: 133-136.
Direct Link - Chern, E.C., Y.L. Tsai and B.H. Olson, 2004. Occurrence of genes associated with enterotoxigenic and enterohemorrhagic Escherichia coli in agricultural waste lagoons. Applied Environ. Microbiol., 70: 356-362.
Direct Link - Grant, M.A., S.D. Weagant and P. Feng, 2001. Glutamate decarboxylase genes as a prescreening marker for detection of pathogenic Escherichia coli groups. Applied Environ. Microbiol., 67: 3110-3114.
Direct Link - Hrudey, S.E. and E.J. Hrudey, 2002. Walkerton and North Battleford-key lessons for public health professionals. Can. J. Public Health, 93: 332-333.
Direct Link - Lantz, P.G., B. Hahn-Hagerdal and P. Radstrom, 1994. Sample preparation methods in PCR-based detection of food pathogens. Trends Food Sci. Technol., 5: 384-389.
Direct Link - Lauber, C.L., L. Glatzer and R.L. Sinsabaugh, 2003. Prevalence of pathogenic Escherichia coli inrecreational waters. J. Great Lakes Res., 29: 301-306.
Direct Link - Martins, M.T., I.G. Rivera, D.L. Clark and B.H. Olson, 1992. Detection of virulence factors in culturable Escherichia coli isolates from water samples by DNA probes and recovery of toxin-bearing strains in minimal O-nitrophenol-β-D-galactopyranoside-4-methylumbelliferyl-β-D-glucuronide media. Applied Environ. Microbiol., 58: 3095-3100.
Direct Link - McDaniels, A.E., E.W. Rice, A.L. Reyes, C.H. Johnson, R.A. Haugland and G.N. Stelma, 1996. Confirmational identification of Escherichia coli, a comparison of genotypic and phenotypic assays for glutamate decarboxylase and β-D-glucuronidase. Applied Environ. Microbiol., 62: 3350-3354.
Direct Link - McLellan, S.L., 2004. Genetic diversity of Escherichia coli isolated from urban rivers and beach water. Applied Environ. Microbiol., 70: 4658-4665.
Direct Link - Naravaneni, R. and K. Jamil, 2005. Rapid detection of food-borne pathogens by using molecular techniques. J. Med. Microbiol., 54: 51-54.
Direct Link - Obi, C.L., E. Green, P.O. Bessong, B. Villiers, A.A. Hoosen, E.O. Igumbor and N. Potgieter, 2004. Gene encoding virulence markers among Escherichia coli isolates from diarrhoeic stool samples and river sources in rural Venda communities of South Africa. Water SA., 30: 37-42.
Direct Link - Smith, D.K., T. Kassam, B. Singh and J.F. Elliot, 1992. Escherichia coli has two homologous glutamate decarboxylase genes that map to distinct loci. J. Bacteriol., 174: 5820-5826.
Direct Link - Sockett, P.N., 1991. The economic implications of human Salmonella infection. J. Applied Bacteriol., 71: 289-295.
Direct Link - Stoeckel, D.M. and V.J. Harwood, 2007. Performance, design and analysis in microbial source tracking studies. Applied Environ. Microbiol., 73: 2405-2415.
CrossRefDirect Link - Swaminathan, B. and P. Feng, 1994. Rapid detection of food-borne pathogenic bacteria. Annu. Rev. Microbiol., 48: 401-426.
Direct Link